- Corresponding Author:
- Urmila Joshi
Prin. K. M. Kundnani College of Pharmacy, Jote Joy Building, Rambhau Salgaonkar Road, Cuffe Parade, India. E−mail: urmilajjoshi@hotmail.com
| Date of Submission | 21 August 2012 |
| Date of Revision | 05 March 2013 |
| Date of Acceptance | 07 March 2013 |
| Indian J Pharm Sci 2013;75(2):233-237 |
Abstract
Quercetin is one of the most abundant naturally occurring flavonoid and is associated with a wide range of biological activities, such as antioxidant, antiinflammatory and anticancer activities. However, there are multiple problems associated with the bioavailability of quercetin, thereby restricting its use. Taking this into consideration, the structure of quercetin was modified by removal of multiple hydroxyl groups and introduction of substituents such as Cl, OCH 3 and N (CH 3 ) 2 on the p-position of the B-ring. The effect of structural modification on the anticancer activity was studied using four different cell lines, including MCF-7, HepG2, HCT-15 and PC-3. Compound 1a has shown an activity better than quercetin in HepG2 cell lines, whereas 1c and 1e showed significant growth inhibition of the HCT-15 cell lines.
Keywords
Anticancer activity, quercetin, structural modifications
Flavonoids are polyphenolic compounds possessing a diphenylpropane skeleton. They are divided into various subgroups depending upon the number and the position of the substituents. Quercetin is one of the most abundant flavonoids present in fruits and the vegetables [1]. Structurally, it is associated with a wide range of biological activities, such as antioxidant, antiinflammatory and anticancer activities [2−5]. It is present in the form of glycosides. The polyphenolic glycosides are too polar to penetrate the intestinal membranes and hence are not very easily absorbed. Release of the aglycones by the action of the microfloral enzymes is needed for the compounds to become absorbable. Although aglycones are permeable, the bioavailability is low due to poor water solubility and a greater extent of conjugation [6,7].
Glucuronidation, sulphation and methylation of the multiple phenolic groups and identification of up to 23 different conjugates of quercetin have been reported [8]. The plasma protein binding of quercetin is as high as 99.1% [9]. Multiple hydroxyl (OH) groups, particularly when present at 3’ and 4’ positions in the molecule, make it susceptible to chemical and microbial degradation in the colon [10]. Important functionalities for the oxygen scavenging action of flavonoids include either the catechol function or the 2,3−double bond along with the 3−OH group [11,12]. In studies of the binding of quercetin with the model membranes, it was observed that the protective action of the flavonoids at the membrane is due to lipophilic as well as the H−bonding interactions of flavonoids [13,14]. Considering the multiple OH groups present in the structure of quercetin and their effect on the metabolism and bioavailability, we decided to modify the structure by reducing the number of OH groups with a view to reduce the degradation. Our previous results with B−ring substituted flavones devoid of hydroxyl groups were encouraging [15]. In view of the importance of the 2,3−double bond, it was decided to retain it. To explore the effect of hydroxy groups at 3 and 7 positions, we decided to synthesise the flavonols.
Compounds 1a−1e were synthesised by Claisen−Schmidt condensation involving the formation of chalcone intermediates, followed by their cyclization to yield flavonols. Briefly, equimolar quantities of 4−substituted benzaldehyde and 2−hydroxyacetophenone/2,4−dihydroxyacetophenone were stirred at room temperature in the presence of potassium hydroxide until complete consumption of aldehyde. The chalcone thus obtained was cyclized in the presence of sodium hydroxide and hydrogen peroxide to give corresponding flavonol (Scheme 1).

Scheme 1: Synthesis of 2−(4−substituted phenyl)−3−hydroxy − c h r o m e n − 4 − o n e a n d 2 − ( 4 − s u b s t i t u t e d p h e n y l ) − 3 , 7 − dihydroxy−chromen−4−one (1a−e). R = H or OH, R1 = Cl, OCH3 or N (CH3)2; (i) EtOH, 1,4−dioxane, 40% w/v KOH; (ii) 1, 4−dioxane, EtOH, 5.4% w/v NaOH, 35% H2O2.
Adopting the above synthetic scheme, five different analogues of quercetin (1a−1e) were synthesised. Spectral analysis of the synthesised compounds is summarised below. Physicochemical properties of the synthesised compounds are listed in Table 1.
| Comp. No. | R | R1 | Molecular formula | % yield | M.P. (º) | Rf value | Elemental analysis % calculated (% found) | |
|---|---|---|---|---|---|---|---|---|
| C | H | |||||||
| Quercetin | − | − | C15H10O7 | 314−316 | 59.609 (59.587) | 3.334 (3.341) | ||
| 1a | H | Cl | C15H9O3Cl | 69 | 197−198 | 0.71 | 66.150 (66.157) | 3.322 (3.181) |
| 1b | H | OCH3 | C16H12O4 | 67 | 196−198 | 0.63 | 71.635 (71.631) | 4.477 (4.413) |
| 1c | H | N (CH3)2 | C17H15O3N1 | 61 | 190−194 | 0.72 | 72.583 (72.431) | 5.374 (5.391) |
| 1d | OH | OCH3 | C16H12O5 | 66 | 192−196 | 0.69 | 67.605 (67.571) | 4.225 (4.181) |
| 1e | OH | N (CH3)2 | C17H15O4N1 | 69 | 198−200 | 0.70 | 68.677 (68.703) | 5.085 (5.113) |
Table 1: Physicochemical data
Quercetin; Fourier transform infrared spectroscopy (FTIR) (KBr, cm−1): 2715−2897 (aromatic C−H), 1664 (C = O), 3282−3404 (O−H), 1014−1132 (C−O−C); Hydrogen−1 nuclear magnetic resonance (1H NMR) (dimethyl sulfoxide [DMSO]−d6, δ ppm): 6.149−7.641 (m, 5H, aromatic), 7.641 (d, 1H, 2’−H), 7.501 (dd, 1H, 6’−H), 6.857 (d, 1H, 5’−H), 6.370 (d, 1H, 8−H), 6.149 (d, 1H, 6−H), 9.288−12.462 (s, 5H, OH groups); Carbon−13 nuclear magnetic resonance (13C NMR) (DMSO−d6 at 323K, δ ppm): 176.4, 164.4, 161.2, 157.0, 148.2 147.4, 145.5,136.2, 122.5, 116.1, 115.6, 115.7, 98.7, 98.7, 93.8.
Compound 1a; melting point: 197−198º, FTIR (KBr, cm−1): 2812−2875 (aromatic C−H), 1614 (C=O), 3284 (O−H), 1091−1107 (C−O−C); 1H NMR (DMSO−d6, δ ppm): 9.63(s), 8.26(s), 8.24(s), 8.12(d), 7.81(t), 7.74(d), 7.64(s), 7.62(s), 7.49(t); 13C NMR (DMSO−d6 at 323K, δ ppm): 173.4, 155.0, 144.5, 139.6, 134.9, 134.1, 130.6, 130.6, 129.7, 129.7, 129.0, 125.2, 125.0, 121.7, 118.8.
Compound 1b; FTIR (KBr, cm−1): 2814−2892 (aromatic C−H), 1612 (C=O), 3204 (O−H), 1087−1111 (C−O−C); 1H NMR (DMSO−d6, δ ppm): 9.26(s), 8.21(s), 8.19(s), 8.12(d), 7.79(t), 7.74(d), 7.47(t), 7.15(s), 7.13(s), 3.87(s); 13C NMR (DMSO−d6 at 323K, δ ppm): 173.2, 160.9, 154.9, 146.1, 138.5, 133.9, 129.8, 125.1, 125.1, 124.9, 121.8, 118.6, 114.5, 114.5, 56.0.
Compound 1c; FTIR (KBr, cm−1): 2810−2902 (aromatic C−H), 1653 (C=O), 3290 (O−H), 1092−1123 (C−O−C), 1232 (C−N); 1H NMR (DMSO−d6, δ ppm): 2.976 (s, 6H, −N(CH3) 2), 6.945−8.219 (m, 8H, aromatic), 8.821 (s, 1H, −OH).
Compound 1d; FTIR (KBr, cm−1): 2813−2890 (aromatic C−H), 1610 (C=O), 3211 (O−H), 1084−1111 (C−O−C); 1H NMR (DMSO−d6, δ ppm): 3.806 (s, 3H, −OCH3), 6.992−7.876 (m, 7H, aromatic), 8.299 (s, 1H, 3−OH), 12.595 (s, 1H, 7−OH).
Compound 1e; FTIR (KBr, cm−1): 2810−2875 (aromatic C−H), 1626 (C=O), 3186 (O−H), 1081−1111 (C−O−C), 1228 (C−N); 1H NMR (DMSO−d6, δ ppm): 2.745 (s, 6H, −N(CH3)2), 6.348−7.489 (m, 7H, aromatic), 8.670 (s, 1H, 3−OH), 12.895 (s, 1H, 7−OH).
The cell lines were grown in Roswell Park Memorial Institute (RPMI) 1640 medium containing 10% foetal bovine serum and 2 mM L−glutamine. For the screening experiment, cells were inoculated into 96 well microtiter plates in 100 μl at plating densities as shown in the study details above, depending on the doubling time of individual cell lines. After cell inoculation, the microtiter plates were incubated at 37°, 5% CO2, 95% air and 100% relative humidity for 24 h prior to addition of experimental drugs.
After 24 h, one 96 well−plate containing 5×103 cells/ well was fixed in situ with trichloroacetic acid (TCA), to represent a measurement of the cell population at the time of drug addiction (time zero [Tz]). Experimental drugs were initially solubilized in DMSO at 100 mg/ml and diluted to 1 mg/ml using water and stored frozen prior to use. At the time of drug addition, an aliquote of frozen concentrate (1 mg/ml) was thawed and diluted to 100, 200, 400 and 800 μg/ml with complete medium containing test article. Aliquots of 10 μl of these different drug dilutions were added to the appropriate microtiter wells already containing 90 μl of medium, resulting in the required final drug concentrations i.e., 10, 20, 40, 80 μg/ml [16].
After the addition, plates were incubated at standard conditions for 48 h and the assay was terminated by the addition of cold TCA. Cells were fixed in situ by the gentle addition of 50 μl of cold 30% (w/v) TCA (final concentration, 10% TCA) and incubated for 60 min at 4°. The supernatant was discarded; plates were washed 5 times with tap water and air dried. Sulforhodamine B solution (50 μl) at 0.4% (w/v) in 1% acetic acid was added to each of the wells and plates were incubated for 20 min at room temperature. After staining, unbound dye was recovered and the residual dye was removed by washing 5 times with 1% acetic acid. The plates were air dried. Bound stain was subsequently eluted with 10 mM trizma base and the absorbance was read on a plate reader at a wavelength of 540 nm with 690 nm reference wavelength. Per cent growth was calculated on a plate−by−plate basis for test wells relative to control wells and expressed as the ratio of average absorbance of the test well to the average absorbance of the control wells multiplied by 100 [17].
Using the six absorbance measurements (Tz, control growth [C] and test growth in the presence of drug at the four concentration levels [Ti]), the percentage growth was calculated at each of the drug concentration levels. Percentage growth inhibition was calculated as: [(Ti-Tz)/(C-Tz)] x 100 for concentrations for which Ti>/=Tz (Ti-Tz) positive or zero and [(Ti-Tz)/Tz] x 100 for concentrations for which Ti<Tz. (Ti-Tz) negative. The results of the anticancer activity of these compounds are shown in Fig. 1.

Fig. 1: Effect of synthesized flavonols on cell growth of different human cancer cell lines. Effect of synthesized flavonols at a concentration of 80 μg/ml on cell growth of different human cancer cell lines. All values are expressed as mean±SD of at least three independent experiments. 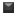 PC-3
PC-3  HCT-15
HCT-15  HEPG2
HEPG2 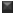 MCF-7.
MCF-7.
It was observed that the presence of an additional OH group at C−7 position of the flavonol increased the time required for the chalcone formation as well as the cyclization. The yields of the 7−OH compounds were low as compared to compounds devoid of 7−OH substitution. The formation of the substituted flavonol was confirmed by NMR spectroscopy, where the compounds showed the peak of the enolic OH at 8.25 and 7/8 protons in the aromatic region at 6.96−7.69. The phenolic OH when present showed an exchangeable peak at 12.6 confirming its presence. The elemental analysis was found within satisfactory limits.
The anticancer testing was carried out on four different cell lines, viz. MCF−7, PC−3, HepG2 and HCT−15, which represent the breast cancer, prostate cancer, hepatic cancer and colon cancer cell lines respectively. Breast, prostate and colon cancers represent the cancers of different tissue origin [18] and the corresponding cell lines were therefore selected. The additional choice of HepG2 cell line was based on the literature report that quercetin shows a significant effect on the growth of HepG2 cells [19].
Compound 1a, which is devoid of majority of the OH groups present in quercetin, shows a significant growth inhibitory effect on the HepG2 cell lines. Replacement of the 4’−Cl of 1a by either 4’−OCH3 (1b) or 4’−N(CH3)2 (1c) led to loss of activity. However, more substituents need to be explored before commenting on the structural requirements at 4’−position, which affect the anticancer activity. We have previously reported that 1a and 1b, both are devoid of antioxidant activity when compared to quercetin [20]. It may be concluded that although the presence of multiple OH groups is a requirement for the antioxidant activity of flavonoids, the same may not be true for the anticancer activity of these compounds.
Compound 1a probably acts through a pathway, which does not involve a reduction of oxidative stress. Structural similarity of the molecule 1a to quercetin prompted us to conclude that the growth inhibitory pathway may involve alterations in either p53 or cyclin−dependent kinase or caspase and/or Phosphatidylinositide-3 (PI-3) activity [21−24]. Compound 1b displayed growth inhibitory activity against MCF−7 cell lines, although lower than quercetin. Quercetin is reported [25] to inhibit type II estrogen binding sites (ER II) where estradiol acts as an agonist. Although structural details of binding of quercetin to ER−II is not reported, it probably takes place through H−bonding involving optimally placed phenolic−OH groups as in the case of diethylstilbestrol. Compound 1b may get involved in such H−bonding through 4’−OCH3 group; it lacks the other OH group in its structure. Taking this into consideration, compound 1d and 1e were synthesised, which contain an additional 7−OH group apart from H−bonding partner at 4’−position. These compounds however did not show any growth inhibitory activity against MCF−7 cell lines. A surprising result was higher growth inhibitory effect of compounds 1c and 1e on the HCT−15 cell lines compared with quercetin. Further work needs to be carried out to ascertain the exact role of 7−OH group and its effect on the inhibition of HCT−15 cell lines.
In conclusion, it can be said that out of the five quercetin analogues devoid of multiple phenolic OH groups, one has shown a promising activity against HepG2 cell lines and two compounds have shown an activity better than quercetin in inhibiting the growth of HCT−15 cell lines. Further studies on these compounds may be useful for understanding the structure−activity relationship of anticancer flavonoids.
Acknowledgements
The authors are thankful to AICTE for the sanction of research grant (F.No. 8023/BOR/RID/RPS−165/2007−08). Support of A. Juvekar of ACTREC, H(S) NCB Board and facilities provided by FTNMR National Facility at TIFR are gratefully acknowledged.
References
- Terao J, Piskula M, Yao Q. Protective effect of epicatechin, epicatechingallate, and quercetin on lipid peroxidation in phospholipid bilayers. Arch BiochemBiophys 1994;308:278−84.
- Formica JV, Regelson W. Review of the biology of quercetin and related bioflavonoids. Food ChemToxicol 1995;33:1061−80.
- Havsteen B. Flavonoids, a class of natural products of high pharmacological potency. BiochemPharmacol 1983;32:1141−8.
- Kuo SM. Antiproliferative potency of structurally distinct dietary flavonoids on human colon cancer cells. Cancer Lett 1996;110:41−8.
- Ranelletti FO, Ricci R, Larocca LM, Maggiano N, Capelli A, Scambia G, et al. Growth−inhibitory effect of quercetin and presence of type−II estrogen−binding sites in human colon−cancer cell lines and primary colorectal tumors. Int J Cancer 1992;50:486−92.
- Ng SP, Wong KY, Zhang L, Zuo Z, Lin G. Evaluation of the first−pass glucuronidation of selected flavones in gut by Caco−2 monolayer model. J Pharm PharmSci 2004;8:1−9.
- Zhang L, Lin G, Chang Q, Zuo Z. Role of intestinal first−pass metabolism of baicalein in its absorption process. Pharm Res 2005;22:1050−8.
- Booth AN, Deeds F, Jones FT, Murray CW. The metabolic fate of rutin and quercetin in the animal body. J BiolChem 1956;223:251−7.
- Hollman PC, van Trijp JM, Buysman MN, van der Gaag MS, Mengelers MJ, de Vries JH, et al. Relative bioavailability of the antioxidant flavonoid quercetin from various foods in man. FEBS Lett1997;418:152−6.
- Karakaya S. Bioavailability of phenolic compounds. Crit Rev Food SciNutr 2004;44:453−64.
- Amic D, Davidovic−Amic D, Beslo D, Trinajstic N. Structure−radical scavenging activity relationships of flavonoids. Croat ChemActa 2003;76:55−61.
- van Acker SA, de Groot MJ, van den Berg DJ, Tromp MN, Donné−Op den Kelder G, van der Vijgh WJ, et al. A quantum chemical explanation of the antioxidant activity of flavonoids.Chem Res Toxicol 1996;9:1305−12.
- Sinha R, Gadhwal MK, Joshi UJ, Srivastava S, Govil G. Interaction of quercetin with DPPC model membrane: Molecular dynamic simulation, DSC and multinuclear NMR studies. J Indian ChemSoc 2011;88:1203−10.
- Sinha R, Gadhwal MK, Joshi UJ, Srivastava S, Govil G. Modifying effect of quercetin on model biomembranes: Studied by molecular dynamic simulation, DSC and NMR. Int J Curr Pharm Res 2012;4:70−9.
- Joshi AJ, Gadhwal MK, Joshi UJ, D’Mello P, Sinha R, Govil G. Synthesis of B−ring substituted flavones and evaluation of their antitumor and antioxidant activities. Med Chem Res 2013 (In press).
- Voigt W. Sulforhodamine B assay and chemosensitivity. Methods Mol Med 2005;110:39−48.
- Papazisis KT, Geromichalos GD, Dimitriadis KA, Kortsaris AH. Optimization of the sulforhodamine B colorimetric assay. J Immunol Methods 1997;208:151−8.
- Scambia G, Ranelletti FO, Panici PB, De Vincenzo R, Bonanno G, Ferrandina G, et al. Quercetin potentiates the effect of adriamycin in a multidrug−resistant MCF−7 human breast−cancer cell line: P−glycoprotein as a possible target. Cancer ChemotherPharmacol 1994;34:459−64.
- Kang ZC, Tsai SJ, Lee H. Quercetin inhibits benzo a pyrene−induced DNA adducts in human Hep G2 cells by altering cytochrome P−450 1A1 gene expression. Nutr Cancer 1999;35:175−9.
- Joshi UJ, Gadge AS, D’Mello P, Sinha R, Srivastava S, Govil G. Antiinflammatory, antioxidant and anticancer activity of quercetin and its analogues. Int J Res Pharm Biomed Sci 2011;2:1756−66.
- Chi C, Chang Y, Ou Y, Hsieh C, Lui W, Peng F, et al. Effect of quercetin on the in vitro and in vivo growth of mouse hepatoma cells. Oncol Rep 1997;4:1021−4.
- Ferry DR, Smith A, Malkhandi J, Fyfe DW, deTakats PG, Anderson D, et al. Phase I clinical trial of the flavonoid quercetin: Pharmacokinetics and evidence for in vivo tyrosine kinase inhibition. Clin Cancer Res 1996;2:659−68.
- Kaneuchi M, Sasaki M, Tanaka Y, Sakuragi N, Fujimoto S, Dahiya R. Quercetin regulates growth of Ishikawa cells through the suppression of EGF and cyclin D1. Int J Oncol 2003;22:159−64.
- Shi M, Wang FS, Wu ZZ. Synergetic anticancer effect of combined quercetin and recombinant adenoviral vector expressing human wild−type p53, GM−CSF and B7−1 genes on hepatocellular carcinoma cells in vitro. World J Gastroenterol 2003;9:73−8.
- Xing N, Chen Y, Mitchell SH, Young CY. Quercetin inhibits the expression and function of the androgen receptor in LNCaP prostate cancer cells. Carcinogenesis 2001;22:409−14.