- *Corresponding Author:
- R. Mohapatra
Department of Pharmaceutics, School of Pharmaceutical Sciences, Siksha ‘O’ Anusandhan (Deemed to be University), Bhubaneswar, Odisha 751003, India
E-mail: rajaram.liku@gmail.com
| Date of Received | 04 June 2021 |
| Date of Revision | 22 December 2021 |
| Date of Acceptance | 25 October 2022 |
| Indian J Pharm Sci 2022;84(5):1334-1337 |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
Molecular docking has now become a novel approach for drug discovery in recent years. Computer-aided drug design is an area that is rapidly growing and has seen many successes. Many big pharmaceutical industries, academic resources and research personnel are using the tools for new drug development. In this review, we have discussed briefly different molecular docking methods, software used in the molecular docking process and their application in drug discovery. Molecular docking is a configuration-based virtual tryout in which computer-generated three dimensional structures of small molecules are given the freedom to interact with the target structure in assorted positions, orientation, and conformations.
Keywords
Molecular docking, binding energy, drug discovery, docking programs
In recent years molecular docking has played a crucial task in in silico drug development. The decisive advantage of the tools is that they require less investment in resources and time in comparison to the in vivo lab studies[1-5]. The dry lab, approach predicts the ligand orientation in a complex formed by the ligand itself with proteins or enzymes[6]. The quantification of the interaction is based on the shape and electrostatic interaction of the docked complex. Many docking programs (more than 50) and tools are now in use in the field of drug research and academics[7-11]. The docking programs like AutoDock, AutoDock Vina, FlexX, DOCK, Surflex, GOLD, Glide, ICM, Cdcker, LigandFit, FRED, MCDock, MOE-Dock, LeDock, rDock and UCSF Dock are used. Among this software AutoDock vina, Glide and GOLD are the top-ranking choices with the best scores[12-14]. GOLD and LeDock are commonly preferred to identify the correct ligand binding site[15,16]. According to Wang et al.[17], 90 % of accuracy of the poses was predicted by Glide and GOLD. From literature, it has been found that the enrichment factor obtained from GOLD was higher as compared to Glide in a screening tryout against factor Xa and produced higher enrichment factors than Glide in a virtual screening trial against factor Xa[18]. Meanwhile, in a comparable tryout, the performance of Glide was superior while the same target (Factor Xa) was under consideration. Depending on the experimental poses, some of these programs were also found to be effective in forecasting the Root Mean Square Deviations (RMSDs) ranging from 1.5 to 2 Å. Still, the contemporary docking programs are facing challenges in handling matters of flexible receptor docking, to be specific receptor backbone flexibility[19,20]. The underlying objective of the docking study is to assess, screen and forecast the computational electrostatics associated with the ligand-receptor complex. A typical molecular docking trial involves two distinct steps. In the first step, ligand conformations are sampled following the protein’s active site. In the second step, the ligand conformations are ranked based on a scoring function. Conceptually, reproduction of experimental binding modes should be performed by sampling algorithms and obtained confirmations should also be ranked as per scoring function. This review work has been focused on molecular docking concerning these two perspectives. Docking analysis is the in silico experimental method comprising the study of the interaction between two molecules. Generally, the larger molecule (receptor protein) is designated as a macromolecule and smaller ones are considered as ligands during the study. The following steps are involved in the docking process. In step 1, involves the retirement of Three Dimensional (3D) structure of receptor protein from the Protein Data Bank (PDB) (fig. 1). Afterward, the pre-processing of the obtained 3D structure should be carried out as per parametric availability. Following pre-processing attributes are performed depending on the need i.e filling the missing residues, removal of water molecules, stabilizing the charges and generation of the side chain. In step 2, the interaction site is predicted on the macromolecule (protein). Further, these sites are regarded as active sites (fig. 2). The receptor protein might have one or more than one active sites. Only one of the concerns should be chosen according to the hypothesis of the experiment[21,22]. In step 3, the structures of the ligands are either obtained or sketched. For retrieving purposes, a database like Pub Chem can be used. Tools like Marvin sketch, Chem sketch and Chem draw can be utilized to sketch the ligands followed by pre-processing. The selection criteria of the ligand should strictly adhere to Pfizer’s rule of five proposed by Lipinsky. The rule is vital in the process of screening of lead structure which has to be optimized in a step-wise fashion to increase the activity and selectivity. The rule also ensures the maintenance of the drug-like physicochemical property of the molecule. For a successful discerning of molecules, adherence to two or more of the following rules is mandatory. The no. of hydrogen bond donors should not be more than 5; The no. of hydrogen bond acceptors should not be more than 10; A molecular mass should be less than 500 Dalton; LogP (Octanol-water partition co-efficient) not more than 5 and the range of molar refractivity to be within 35-125. Additional rules has been proposed by Veber with respect to the presence of number of rotatable bonds (should be <10)[23]. Step 4 is the final step of the docking process. The screened and pre-processed ligand molecule is processed in the software for docking against the receptor protein and the molecular interaction between ligand and receptor protein is analyzed (fig. 3). The best-docked ligand complex is characterized based on docking score which is generated by the tools. In the past twenty years, various molecular docking tools have been formulated (Table 1). (Source: Chaudhary KK, Mishra N (2016) A Review on Molecular Docking: Novel Tool for Drug Discovery, (JSM Chem 4 (3): 1029) represents the different molecular docking tools[24]. The resultant binding interaction between ligand and macromolecule in the docking approach may result in the activation or inhibition of the receptor enzyme. Meanwhile, in the case of the receptor proteins and ligand binding may lead to agonism or antagonism. The docking approach can be applied in the field of drug discovery related to target identification through virtual trials; development of potent and selective analogs through optimization; pollutant prediction, which can be combated by enzymes; biological activity prediction; prediction of the active binding site through the blind approach; protein de-orphanization; interaction analysis between protein-protein and protein-nucleic acid; structural and functional analysis; enzyme catalytic reactions and protein engineering. The molecular docking technique presents a significant approach in the area of drug discovery, optimization and analysis. The easy access of structural databases and simple visualization of molecules has now become essential components of the process. Nowadays the rapid expansion of docking software programs (commercial) among the user interfaces has paved the way for easy accessibility. Molecular docking is a computer-aided drug designing tool that relies on software programs[25]. The instance of logic, algorithms is the backbone of the designing tool. With increased input with respect to algorithms from the experts of the field, the method is gaining acceptance in terms of accuracy, sophistication, sensitiveness and perception. Even public domain programs are now equipped with at par functionality with that of the commercial ones. The flawless input commands and crispy graphical representations have improved the aesthetics of the tools, making it an elegant and effective way of drug design. Other than drug discovery, the in silico approach has been employed in formulation development, solubility analysis and drug complex stability study. Now also the technique has been adopted in the field of genomics, proteomics, and computational enzymology.

Fig. 1: (A): 3D structure of receptor protein (6A93) and (B): 3D structure of ligand (Rutin)

Fig. 2: Visualization of 2D models of docked complexes depicted by Discovery Studio Visualizer 3.5, depicting interactions of 5HT2A receptor (6A93) with phyto compound
Note: 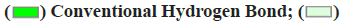 Carbon Hydrogen Bond;
Carbon Hydrogen Bond; 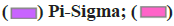 Pi-Pi T shaped and
Pi-Pi T shaped and  Pi-Alkyl
Pi-Alkyl

Fig. 3: Visualization of 3D models of docked complexes depicted by Discovery Studio Visualizer 3.5, depicting interactions of 5HT2A receptor (6A93) with phyto compound
Note: 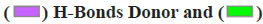 Acceptor
Acceptor
| Software | Company/Designer | Licence terms | Supported platforms | Docking approaches | Scoring function |
|---|---|---|---|---|---|
| Auto Dock | D. S. Good sell and A. J. Olson The Scripps Research Institute | Free for academic use | Unix, Mac OSX, Linux, SGI | Genetic algorithm Lamarckian genetic algorithm simulated annealing | Auto dock (force-field methods) |
| Dock | I. kuntz university of California, san francisco | Free for academic use | Unix, Linux, Sun, IBM AIX, Mac OSX, Windows | Shape fitting (sphere sets) | Chem score GB/SA salvation scoring, other |
| Flex X | T. Lengauer and M. Rarey Bio SolveIT | Commercial Free evaluation (6 w) | Unix, Linux, SGI, Sun Windows | Incremental construction | FlexXscore, PLP, Screen Score, Drug Score |
| FRED | Open eye scientific software | Free for academic use | Unix, Linux, SGI, Mac OSX, IBM AIX, Windows | Shape fitting (Gaussian) | Screen Score, PLP, Gaussian shape score, user defined |
| Glide | Schrodinger Inc. | Commercial | Unix, Linux, SGI, IBM Aix | Monte Carlo Sampling | Glide Score Glide comp |
| GOLD | Cambridge crystallographic data centre | Commercial Free evaluation (2 mo) | Linux, SGI, Sun, IBM, Windows | Genetic algorithm | Gold Score, Chem Score user defined |
| LigandFit | Accelrys Inc. | Commercial | Linux, SGI, IBM AIX | Monte carlo sampling | Lig Score, PLP, PMF |
Table 1: List of Protein-Ligand Molecular Modelling Tools
Acknowledgment:
The authors are grateful to Prof. Manoj Ranjan Nayak president, of Siksha ‘O’ Anusandhan (Deemed to be University) for the support and facilities.
Conflict of interest:
The authors declare no conflict of interest.
References
- Mohapatra R, Mallick S, Nanda A, Sahoo RN, Pramanik A, Bose A, et al. Analysis of steady state and non-steady state corneal permeation of diclofenac. RSC Adv 2016;6(38):31976-87.
- Sahoo RN, Nanda A, Pramanik A, Nandi S, Swain R, Pradhan SK, Mallick S. Interactions between Ibuprofen and Silicified-MCC: Characterization, drug release and modeling approaches. Acta Chim Slov 2019;66(4):923-33.
[Crossref] [Google Scholar] [PubMed]
- Pramanik A, Sahoo RN, Pradhan SK, Mallick S. Characterization and molecular docking of kaolin based cellulosic film for extending ophthalmic drug delivery. Indian J Pharm Sci 2021;83(4):794-807.
- Nanda A, Sahoo RN, Pramanik A, Mohapatra R, Pradhan SK, Thirumurugan A, et al. Drug-in-mucoadhesive type film for ocular anti-inflammatory potential of amlodipine: Effect of sulphobutyl-ether-beta-cyclodextrin on permeation and molecular docking characterization. Colloids Surf B Biointerfaces 2018;172:555-64.
[Crossref] [Google Scholar] [PubMed]
- Dash R, Sahoo RN, Nandi S, Swain R, Mallick S. Sustained release bioadhesive suppository formulation for systemic delivery of ornidazole: In silico docking study. Indian J Pharm Edu Res 2019;53(4):S580-6.
- Lengauer T, Rarey M. Computational methods for biomolecular docking. Curr Opin Struct Biol 1996;6(3):402-6.
[Crossref] [Google Scholar] [PubMed]
- Jorgensen WL. The many roles of computation in drug discovery. Science 2004;303(5665):1813-8.
[Crossref] [Google Scholar] [PubMed]
- Bajorath J. Integration of virtual and high-throughput screening. Nat Rev Drug Discov 2002;1(11):882-94.
[Crossref] [Google Scholar] [PubMed]
- Walters WP, Stahl MT, Murcko MA. Virtual screening—an overview. Drug Discov Today 1998;3(4):160-78.
- Langer T, Hoffmann RD. Virtual screening an effective tool for lead structure discovery. Curr Pharm Des 2001;7(7):509-27.
[Crossref] [Google Scholar] [PubMed]
- Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat Rev Drug Discov 2004;3(11):935-49.
[Crossref] [Google Scholar] [PubMed]
- Potluri H, Prasanth DS, Atmakuri LR. In vivo antinociceptive effect of methanolic extract of Ipomoea marginata Desr. in rodents as well as in silico molecular docking of some phytoconstituents from the plant. Indian J Pharm Sci 2021;83(4):732-41.
- Tomar NR, Singh V, Marla SS, Chandra R, Kumar R, Kumar A. Molecular docking studies with rabies virus glycoprotein to design viral therapeutics. Indian J Pharm Sci 2010;72(4):486.
[Crossref] [Google Scholar] [PubMed]
- Ghode P, Jain SK. Structural Requirements for some 3-amino-N-substituted-4-(substituted phenyl) Butanamides as dipeptidyl peptidase-IV inhibitors using 3D-QSAR and molecular docking approaches. Indian J Pharm Sci 2018;79(6):974-86.
- Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein–ligand docking using GOLD. Proteins 2003;52(4):609-23.
[Crossref] [Google Scholar] [PubMed]
- Liu N, Xu Z. Using LeDock as a docking tool for computational drug design. IOP Conference Series: Earth and Environmental Science 2019;218(1):012143. IOP Publishing.
- Wang Z, Sun H, Yao X, Li D, Xu L, Li Y, et al. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: The prediction accuracy of sampling power and scoring power. Physical Chem Chem Phys 2016;18(18):12964-75.
[Crossref] [Google Scholar] [PubMed]
- Yousuf M, Shaikh NN, Ul-Haq Z, Choudhary MI. Bioinformatics: A rational combine approach used for the identification and in vitro activity evaluation of potent β-Glucuronidase inhibitors. PloS One 2018;13(12):e0200502.
[Crossref] [Google Scholar] [PubMed]
- Bissantz C, Folkers G, Rognan D. Protein-based virtual screening of chemical databases. 1. Evaluation of different docking/scoring combinations. J Med Chem 2000;43(25):4759-67.
[Crossref] [Google Scholar] [PubMed]
- Dixon JS. Evaluation of the CASP2 docking section. Proteins 1997;29(1):198-204.
[Crossref] [Google Scholar] [PubMed]
- Mcmartin C, Bohacek RS. QXP: powerful, rapid computer algorithms for structure-based drug design. J Comput Aided Mol Des 1997;11(4):333-44.
[Crossref] [Google Scholar] [PubMed]
- Schnecke V, Kuhn LA. Virtual screening with solvation and ligand-induced complementarity. In Virtual Screening: An alternative or complement to high throughput screening? 2000:171-190.
- Pradeep Singh S, Kumar Konwar B. Virtual screening and molecular descriptor analysis on dietary phytochemicals against heat shock protein 90 enzyme. Lett Drug Des Discov 2014;11(1):40-9.
- Chaudhary KK, Mishra N. A review on molecular docking: Novel tool for drug discovery. Databases 2016;3(4):1029.
- López-López E, Bajorath J, Medina-Franco JL. Informatics for chemistry, biology, and biomedical sciences. J Chem Inf Model 2020;61(1):26-35.
[Crossref] [Google Scholar] [PubMed]





