- *Corresponding Author:
- Anita Lalwani
K. B. Institute of Pharmaceutical Education and Research, Gh-6 Road, Sector-23 Gandhinagar - 382 023, India
E-mail: lalwanianitain@yahoo.com
| Date of Submission | 28 April 2006 |
| Date of Revision | 28 May 2007 |
| Date of Acceptance | 4 July 2007 |
| Indian J Pharm Sci 2007, 69 (4): 489-497 |
Abstract
The ability to deliver therapeutic agents to a patient in a pulsatile or staggered release profile has been a major goal in drug delivery research recently. This review will cover methods that have been developed to control drug delivery profile with different polymeric systems. Externally and internally controlled systems will be considered, including a range of technologies like preprogrammed systems as well as systems that are sensitive to modulated enzymatic or hydrolytic degradation, pH, magnetic fields, ultrasounds, electric fields, temperature, light and mechanical stimulation. These systems have the potential to improve the quality of life for patients undergoing therapy with a variable dosing regime.
While newer and more powerful drugs continue to be developed, increasing attention is being given to the methods by which these active substances are administered. A new development, polymeric controlled drug delivery, has evolved from the need for prolonged and better control of the drug administration. The controlled-release devices, which are already available commercially, can maintain the drug in the desired therapeutic range with just a single dose, localize delivery of the drug in to a particular body compartment, which lowers the systemic drug level, reduces the need for follow-up care, preserves medications that are rapidly destroyed by the body, and increases patient comfort and/or patient compliance. The basic approach that drug concentration-effect relationships are significantly invariant as a function of time in man has led to the development of constant rate drug delivery systems. Nevertheless, there are a number of clinical situations where such an approach may not be sufÞ cient. These include delivery of insulin for patients with diabetes mellitus, antiarrhythmics for patients with heart rhythm disorders, gastric acid inhibitors for ulcer control, nitrates for patients with angina pectoris, as well as selective β-blockade, birth control, general hormone replacement, immunization, and cancer chemotherapy. Recent studies in the field of chronopharmacology indicate that the onset of certain diseases exhibit strong circadian temporal dependency. Thus, drug delivery patterns can be further optimized by pulsed or self-regulated delivery, adjusted to the staging of biological rhythms.
Pre-Programmed Drug Delivery System
These systems are designed to release drug in pulses governed by the device fabrication and ideally, independent of the environment. The release mechanisms employed include bulk erosion of the polymer in which drug release by diffusion is restricted, surface erosion of layered devices composed of alternating drug-containing and drug-free layers, osmotically controlled rupture and enzymatic degradation of liposomes.
Bulk-eroding systems
Poly(lactide-co-glycolide) PLGA is a bulk-eroding polymer, where the ingress of water is faster than the rate of degradation. In this case, degradation takes place throughout the polymer sample, and proceeds until a critical molecular weight is reached, at which point degradation products become small enough to be solubilised. At this point, the structure starts to become significantly more porous and hydrated, and it is possible for drug dissolved in the polymer matrix to be released, corresponding to the time required for critical molecular weight to be reached. Hence there is a time lag before the drug can be released, corresponding to the time required for critical molecular weight to be reached (fig. 1). A number of research groups have investigated microsphere formulations of these polymers [1-8]. Microspheres can be formulated as subcutaneous injections and produce a depot of antigen in the subcutaneous site. Pulsatile release of antigens can be achieved by administration of a single injection containing two or more types of microspheres that have different release profiles [9].

Figure 1: Theoretical pulsatile release of a drug from a bulk eroding
polymer system.
The time between initial release and booster release is determined
by the period of bulk erosion to reach a molecular weight that causes
porosity in the matrix. MW is molecular weight
Alonso and coworkers developed a controlled delivery system for tetanus toxoid vaccine. PLGA with a 50:50 lactide:glycolide ratio was used to form microspheres, with drug dissolved in the polymer. In vitro studies show a sigmoidal release profile, with an increase in release rate after 15 days [10].
Vaccines are proteinous in nature and so a common problem, when matrix type delivery systems are used for vaccines, is their degradation. The factors causing degradation include: exposure to moisture inside the delivery system, an acidic environment caused by the polymer degradation products and the possibility of reaction with these degradation products. In order to limit these problems, the drug is encapsulated in a reservoir microcapsule, with the drug being protected by suspension in oil. Sanchez and coworkers investigated ways of avoiding denaturation of vaccine. Work was carried out on two different PLGA compositions, 50:50 and 75:25 lactide:glycolide. In both cases, the majority of drug release occurred in pulse, with a time delay of 3 weeks for 50:50 composition and 7 weeks for the 75:25 composition. As in the matrix system, drug could only be released from the reservoir when a critical level of the porosity was reached in the membrane. This explains why the 50:50 polymer which has a faster degradation rate, gave an earlier release. The researchers suggested an injectable ‘cocktail’ formulation, with a mixture of a priming dose of tetanus antigen, 50:50 microcapsules releasing a booster at 3 weeks, and another booster at 7 weeks, from the 75:25 microcapsules [11].
Surface-eroding systems
Certain polymers such as poly(ortho) esters and polyanhydrides, degrade by surface erosion. This means that the rate of degradation of the polymer is such that mass loss is faster than the ingress of water into the bulk. Hence the sample is eroded from the surface, at a controlled and predictable rate. Drug dissolved in the polymer is released at a constant rate as erosion progresses, provided that the device surface area does not change. As drug release begins as soon as polymer erosion starts (which is generally almost immediate), it is difficult to produce a pulsatile system using a single surface-eroding polymer. Therefore, most devices involve a layered structure. In theory, pulsatile release would occur if the drug release, by diffusion, through adjacent layers is restricted and drug-free layers are placed between drug-containing layers (fig. 2).

Figure 2: Theoretical pulsatile release of a drug from surface-eroding
polymeric systems.
The time between initial release and booster release is determined
by the erosion of the drug-free layer.
A technique that has successfully demonstrated sequential release of several model drugs is the fabrication of a multilayered polymer matrix with the alternating drug containing and spacer layers. As seen in fig 3, the matrix is commonly surrounded by an impermeable shell, permitting release of the entrapped drug only during degradation of the polymer matrix. For this to occur, the polymer must be vulnerable to hydrolysis or biodegradation by a component in the surrounding media. A small amount of drug is often released via diffusion through the inactive matrix, but this is usually negligible in comparison to the amount of drug released as the active matrix undergoes erosion. The length of the active and inactive delivery phases can be altered by changing the type and quantity of the isolating layer, the type and quantity of the drug containing layer and the type of the drug that is being released.

Figure 3: Schematic representation of multilayered pulsatile delivery
systems
A. Multilayered pulsatile delivery systems with one face exposed
to the local environment [14], and B. Cylindrical multilayered delivery
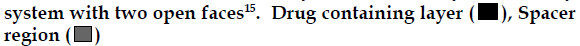
As an example to this type of system, a research group was able to release three pulses of brilliant blue with inactive phases ranging from 20-110 min using different amounts and types of polyanhydrides as the isolating layers and poly[(ethylglycinate)(benzylamin o acethydroxamate)phosphazene] (PEBP) as the drug containing layer. For this application, polyanhydrides and PEBP layers were compression molded to form a multilayered cylindrical core, which was coated with poly(lactide-co-1,3-trimethylene carbonate) film over all the surfaces except for one of the device. The hydrolysis of the PEBP is highly dependent on the pH of the surrounding media, dissolving much more rapidly under neutral and basic conditions as oppose to acidic conditions [12]. The degradation products of polyanhydrides create an acidic environment within the delivery device, preventing the hydrolysis of PEBP and resultant drug release until the entire polyanhydride has been eroded [13-15].
Gopferich also investigated a multilaminate matrix device. A core of drug-filled polyanhydride was surrounded by a drug-free poly(lactide-co-glycolide) PLGA layer, then a second drug-loaded coating of polyanhydride was done. In vitro measurements showed a two-phase drug release, with an initial release from the outer coating on first day followed by a second phase after 2 weeks [16-20].
Osmotically controlled system
Increasing the osmotic pressure within a device may be used as driving force to activate pulsatile release [21]. The principle of an osmotically bursting delivery system is shown in fig 4. Antigen was included in a compressed core of Explotab® which was coated with Eudragit® S film plasticized with dibutyl phthalate and Eudragit® NE with 3% hydroxypropylmethylcellulose (HPMC) as pore former formed an outer coat. When this system came in contact with an aqueous environment, HPMC in the outer coat dissolved to create pores through which water could access Eudragit S film and then enter the implant core. The compressed core would then swell until Eudragit S film ruptured and antigen was released in a single shot. In vitro release profile showed a delay of between 14 and 26 days before release of model antigen. Pulsatile antigen administration was achieved by coadministration of coated and uncoated tablets and produced elevated antibody titers for at least 3 months. Cardamone et al. used osmotic pressure to achieve pulsatile release of tetanus toxoid [22].

Figure 4: Theoretical pulsatile release of a drug from an osmotically
driven system.
The time until the booster release is determined by the inß ux of
water into and swelling of the tablet core. Gray region indicates
the pore formation and subsequent uptake of water occurs through
these pores
Enzymatically-activated liposomes
Liposomes have been used as drug delivery carriers because of their structural similarity to cell membranes. However, uptake by the reticuloendothelial system or a destabilization due to the absorption of plasma proteins on the lipid bilayer have been the major drawbacks and have thus limited the application of these formulations. To overcome the drawbacks of liposomes as a drug carrier system, Langer and co-workers [23,24] incorporated drug-loaded liposomes into microcapsules of alginate hydrogels. The hydrogel matrix was designed to protect the liposomes from degradation and/or dispersion in vivo, as well as possibly regulate the release rate of incorporated drug molecules. To achieve a pulsatile release of drug molecules the liposomes inside the microcapsules were coated with phospholipase A2. Phospholipase A2 was shown to gather at the water/liposome interfaces and remove an acyl group from the phospholipids in the liposome. Destabilised liposomes release their drug molecules from the interior, thus allowing drug release to be regulated by the rate determining microcapsule memberane (fig. 5).

Figure 5: Schematic representation of liposome-loaded alginate-poly(llysine) microcapsules.
Closed-loop delivery systems
Closed-loop delivery systems are those that are self-regulating. They are similar to the programmed delivery devices in that they do not depend on an external signal to initiate drug delivery. However, they are not restricted to releasing their contents at predetermined times. Instead, they respond to changes in local environment, such as the presence or absence of a specific molecule (fig. 6).

Figure 6: Theoretical pulsatile release from a triggered-system.
Glucose-sensitive systems
There has been much interest in the development of stimuli-sensitive delivery systems that release a therapeutic agent in presence of specific enzyme or protein. One prominent application of this technology has been development of a system that can autonomously release insulin in response to elevated blood glucose levels. Several existing strategies that may be feasible for glucose-responsive drug delivery are discussed below:
pH-dependent systems for Glucose stimulated drug delivery are based on the reaction that glucose oxidase catalyses oxidation of glucose to gluconic acid. This reaction can be used to drive the swelling of pH-dependent membrane. A dual membrane system was formed. In the first membrane, glucose oxidase was immobilized on cross linked polyacrylamide and this was referred to as glucose sensing membrane. Co-polymer membrane composed of N,Ndiethylaminoethyl methacrylate and 2-hydroxypropyl methacrylate (DEA-HPMA) formed the barrier membrane and worked as an interface between insulin reservoir and sensing membrane [25].
As shown in fig. 7, gluconic acid formed by the interaction of glucose and glucose oxidase, caused the tertiary amine groups in the barrier memberane to protonate and induce a swelling response in the memberane. Insulin in the reservoir was able to diffuse across the swollen barrier memberane. When the glucose concentration decreased, the pH of the barrier memberane increased and it returned to a more collapsed and impermeable state.

Figure 7: Schematic diagram of a glucose-sensitive dual-membrane
system.
(A) Diagram of a glucose-sensitive dual-membrane system. (B)
The membrane bordering the release media responds to increased
glucose levels by increasing the permeability of the membrane
bordering the insulin reservoir [25].
Another glucose - sensitive delivery system is based on the competitive binding of concanavalin (con A), which is a glucose binding lectin. Obaidat and Park prepared a copolymer of acrylamide and allyl glucose. The side chain glucose units in the copolymer were bound to con A. These hydrogel showed a glucose responsive, sol-gel phase transition, depending on the external glucose concentration due to the competition between free glucose and con A. Con A acts as cross linker for the polymer chains due to the presence of four glucose-binding sites on the molecule, but competitive binding with glucose disrupts these cross links, making the material more permeable and thus increasing the rate of drug delivery (fig. 8). Membranes and hydrogels consisting of these copolymers can act as gates or depots for glucosedependent insulin delivery [26,27].

Figure 8: Sol–gel phase transition in polymers cross linked with Con
A. [27]
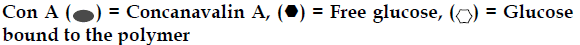
Open-Loop Delivery Systems
Open-loop delivery systems are not self-regulating, but instead require externally generated environmental changes to initiate drug delivery. These can include magnetic fields, ultrasound, electric field, temperature, light and mechanical force.
Magnetic field
Use of an oscillating magnetic field to modulate the rates of drug delivery from a polymer matrix was one of the first methodologies investigated to achieve an externally controlled drug delivery system [28]. Magnetic carriers can receive their magnetic response to a magnetic field from incorporated materials such as magnetite, iron, nickel, cobalt and steel.
Magnetic steel beads were embedded in an ethylene and vinyl acetate (EVAc) copolymer matrix that was loaded with bovine serum albumin as a model drug. Authors demonstrated increased rates of drug release in the presence of an oscillating magnetic field [29]. During exposure to the magnetic field, the beads oscillate within the matrix, alternatively creating compressive and tensile forces. This in turn acts as a pump to push an increased amount of the drug molecule out of the matrix. Co-polymers with a higher Young’s modulus were more resistant to the induced motion of steel beads, and consequently the magnetic field has less effect on the rate of drug release from these materials [30].
Saslawski et al. [31] developed different formulations for in vitro magnetically triggered delivery of insulin based on alginate spheres. In an experiment, ferrite microparticles (1 μm) and insulin powder were dispersed in sodium alginate aqueous solution. The ferrite-insulin alginate suspension was later dropped in aqueous calcium chloride solution which causes the formation of cross linked alginate spheres, which were further cross linked with aqueous solution of poly(L-lysine) or poly(ethylene imine). They described that the magnetic field characteristics due to the ferrite microparticles and the mechanical properties of the polymer matrices could play role in controlling the release rates of insulin from the system.
US Patent 2006997863 [32] provides a treatment method that involves the administration of a magnetic material composition, which contains single-domain magnetic particles attached to a target-specific ligand, to a patient and the application of an alternating magnetic field to inductively heat the magnetic material composition, which cause the triggered release of therapeutic agents at the target tumor or cancer cells.
Ultrasound
Ultrasound is mostly used as an enhancer for the improvement of drug permeation through biological barriers, such as skin, lungs, intestinal wall and blood vessels. There are several reports describing the effect of ultrasound on controlled drug delivery [33-39]. Kost et al. described an ultrasound-enhanced polymer degradation system. During polymer degradation incorporated drug molecules were released by repeated ultrasonic exposure. As degradation of biodegradable matrix was enhanced by ultrasonic exposure, the rate of drug release also increased. Thus, pulsed drug delivery was achieved by the on-off application of ultrasound [40]. Supersaxo et al. [41] also reported macromolecular drug release from biodegradable poly (lactic acid) microspheres. Drug release from porous poly (lactic acid) microspheres showed an initial burst followed by a sustained release for over several months. When ultrasound was applied to this release system, pulsatile and reversible drug release was observed. Authors speculated that ultrasonic exposure resulted in the enhancement of water permeation within microspheres of the polymer matrix, inducing drug dissolution into the releasing media.
Miyazaki et al. [42] used ultrasound to achieve up to a 27-fold increase in the release of 5-fluorouracil from an ethylene and vinyl acetate (EVAc) matrix. Increasing the strength of the ultrasound resulted in a proportional increase in the amount of 5-ß uorouracil released.
Increase in the rate of p-nitroaniline delivery from a polyanhydride matrix during ultrasonic irradiation is reported [43]. The authors noted that the increase in drug delivery was greater than the increase in matrix erosion when the ultrasound triggering was active. Thus it was hypothesized that acoustic cavitation by ultrasonic irradiation was responsible for the modulated delivery of p-nitroaniline [44].
Temperature
Temperature is the most widely utilized triggering signal for a variety of triggered or pulsatile drug delivery systems. The use of temperature as a signal has been justified by the fact that the body temperature often deviates from the physiological temperature (37º) in the presence of pathogens or pyrogens. This deviation sometimes can be a useful stimulus that activates the release of therapeutic agents from various temperature-responsive drug delivery systems for disease accompanying fever. Thermal stimuli-regulated pulsed drug release is established through the design of drug delivery devices such as hydrogels and micelles.
Thermo-responsive hydrogel systems use hydrogels that undergo reversible volume changes in response to changes in temperature. These gels shrink at a transition temperature that is related to the lower critical solution temperature (LCST) of the linear polymer from which the gel is made. Thermo-sensitive hydrogels have a certain affinity for water, and thus swell at temperatures below the transition temperature, whereas they expel water and thus shrink or “deswell” at temperatures above the transition temperature. Thermally-responsive hydrogels and membranes have been extensively evaluated as platforms for the pulsatile delivery of drugs [45].
Special attention has been given to the thermally responsive poly(N-isopropylacrylamide) and its derivative hydrogels. Poly(N-isopropyl acryl amide) (PIPAAm) cross-linked gels have shown thermo responsive, discontinuous swelling/deswelling phases: swelling for example, at temperatures below 32°, while shrinking above this temperature. A sudden temperature increase above the transition temperature of these gels resulted in formation of a dense, shrunken layer on the gel surface (skin layer), which hindered water permeation from inside the gel into the environment. Drug release from the PIPAAm hydrogels at temperature below 32° was governed by diffusion, while above this temperature drug release was stopped completely, due to the ‘skin layer’ formation on the gel surface (on-off drug release regulation) [46-49].
Kaneka and co-workers developed a new method to accelerate gel swelling/deswelling kinetics based on molecular design of gel structure. Free mobile linear PIPAAm chains were grafted within the cross-linked PIPAAm hydrogels (fig. 9). These gels had the same transition temperature as conventional PIPAAm gels and existed in the swollen state below the transition temperature, while above this temperature they shrank. PIPAAm grafted gels showed rapid deswelling kinetics without formation of skin layer on surface. This is probably due to rapid dehydration of graft chains formed by hydrophobic aggregation on the three-dimensional cross-linked chains [50-52]. A similar rapid deswelling phase was achieved by incorporating poly(ethylene glycol) graft chains in PIPAAm cross linked hydrogels [53].

Figure 9: Model structure of freely mobile linear PIPAAm-grafted
PIPAAm hydrogels.
PIPAAm = Poly(N-isopropylacrylamide)
Yuk et al. designed temperature-sensitive drug delivery systems using a mixture of poly(ethylene oxide)- poly(propylene oxide)-poly(ethylene oxide) triblock copolymer (F-68) and poly vinyl alcohol (PVA). Change in the ratio of F-68/PVA could be used to manipulate the swelling transition of polymer complex gel. Authors demonstrated pulsatile release of acetaminophen in response to pulsatile change in temperature between 35 and 40° [54].
US Patents 6733788 [55] and 20020015712 [56] describe a medical device containing thermo-sensitive cellulose gel structure, which can deliver the bioactive solute compounds to a target location in the body. The gel structure deswells at certain temperature and expels the biologically active solute with an increase in gel temperature. All of the loaded solute released in a relatively short period of time under the inß uence of increased temperature of the body.
Thermo-responsive polymeric micelle systems constitute polymeric micelles whose properties and biological interests make them a most noteworthy candidate as drug carrier for the treatment of cancer [57]. The polymeric micelle is composed of amphiphilic block copolymers exhibiting a hydrophobic core with a hydrophilic corona (fig. 10). Due to these unique characteristics, polymer micelles exhibit stealth characteristics and are not detected by the body defense system (reticuloendothelial system). Thus passive targeting could be achieved through enhanced permeation retention (EPR) effect of tumor sites [58]. Okano and coworkers used an end functionalized PIPAAm to prepare block copolymers. Hydrophobic polymers, such as poly(butyl methacrylate) (PBMA), polystyrene (PSt) [59,60, poly(lactic acid) (PLA) [61,62] were used. Block copolymers formed micellar structure (with core-shell structure) in aqueous solution below PIPAAm’s transition temperature. The shell was made from thermo-responsive PIPAAm, while the core consisted of hydrophobic polymer aggregates of poly(butyl methacrylate) (PBMA). The hydrated PIPAAm corona acted as an inert material towards all biological entities below PIPAAm’s LCST. However, upon temperature increase above 32° hydrated PIPAAm chains became hydrophobic, due to dehydration of polymer chains, thus resulting in aggregation and precipitation. The hydrophobic anticancer drug, andriamycin, was incorporated in to PBMA micelle cores. At temperatures below PIPAAm’s low crystalline solution temperature (LCST), drug release was at a minimum, with a value less than 10%. However upon temperature increase above PIPAAm’s LCST, accelerated release of andriamycin was observed.

Figure 10: Structure of block copolymer micelles.
PIPAAm = Poly(N-isopropylacrylamide), PBMA = Poly(butyl
methacrylate), PLA = Poly(lactic acid)
Electric field
An electric field as an external stimulus has advantages such as availability of equipment, which allows precise control with regards to the magnitude of the current, duration of electric pulses, interval between pulses etc. Electrically responsive delivery systems are prepared from polyelectrolytes (polymers which contain relatively high concentration of ionisable groups along the backbone chain) and are thus pHresponsive as well as electro responsive. Under the inß uence of electric field, electro responsive hydrogels generally deswell, swell or erode. The mechanisms of drug release include ejection of drug from the gel as the fluid phase synereses out, drug diffusion along a concentration gradient, and electrophoresis of charged drug towards an oppositely charged electrode and liberation of the entrapped drug as the gel complex erodes [63]. Synthetic as well as naturally occurring polymers, separately or in combinations, have been used for this purpose. Examples of naturally occurring polymers include hyaluronic acid, chondrotin sulphate, agarose, carbomer, xanthan gum and calcium alginate. The synthetic polymers are generally acrylate and methacrylate derivatives such as partially hydrolysed polyacrylamide, polydimethylaminopropyl acrylamide.
Poly(2-acrlamide-2-methylpropanesulfonic acid-cobutyl methacrylate) (P(AMPS-co-BMA) hydrogels were used for electric stimuli-induced drug delivery system [64-66]. Positively charged edrophonium chloride was incorporated as drug molecule within negatively charged P(AMPS-co-BMA) hydrogels. By applying an electric field, ion exchange between edrophonium ions and protons commenced at cathode, resulting in rapid drug release from hydrogels. This rapid drug release was attributed to the electrostatic force, squeezing effect, and electro-osmosis of the gel. Complete on-off drug release was achieved, as no drug release was apparent without the application of electric current.
Complex multi-component gels or interpenetrating networks have been prepared in order to enhance the gel’s electroresponsiveness [67]. Lee and coworkers prepared calcium alginate/ poly(acrylic acid) composites, where the polyacrylic acid (PAA) chains were expected to be entangled through the calcium alginate matrix. PAA, which contains a large number of free carboxylic groups, was included to increase the gel’s sensitivity to pH and electrical stimuli. The increased proportion of PAA in the composites led to a greater pH and electro-response [68].
Light
The interaction between light and material can be used to modulate drug delivery. This can be accomplished by combining a material that absorbs light at a desired wavelength and a material that uses energy from the absorbed light to modulate drug delivery. Gold nanoshells are a new class of optically active nanoparticles that consist of a thin layer of gold surrounding a core. The optical properties of the nanoshells can be tuned over the visible and near IR spectrum. Embedding the nanoshells in a NIPAAm-co-AAM hydrogel formed the required composite material. When exposed to near-infrared light, nanoshells absorb the light and convert it to heat, raising the temperature of composite hydrogel above its LCST. The hydrogel collapses and this results in an increased rate of release of soluble drug held with in the matrix [69,70] (fig. 11).

Figure 11: A representative nanoshell-composite hydrogel in the fully swollen and collapsed states
Mechanical force
Drug delivery can also be initiated by mechanical stimulation of an implant. Alginate hydrogels that release vascular endothelial growth factor in response to compressive forces of varying strain amplitudes were developed. Free drug that is held with in the polymer matrix is released during compression; once the strain is removed hydrogel returns to its original volume. This concept is essentially similar to squeezing the drug out of a sponge [71].
Conclusions
A significant amount of progress has been made towards achieving pulsatile drug delivery systems that can effectively treat diseases with non-constant dosing therapies, such as diabetes. Products that are currently under development for commercialization are for the delivery of proteins, hormones, pain medications, and other pharmaceutical compounds. Some of the current programmable systems that employ polymerbased drug delivery include Covera HS, Verelan PM, Cardizem LA, Innopran XL, Uniphyl and naproxen sodium from Andrx Pharmaceuticals. The major drawbacks arise from the biological variations among individuals. The key considerations in the design of polymer based pulsatile systems are the biocompatibility and the toxicity of the polymers used, response to the external stimuli, the ability to maintain the desired levels of drugs in serum, the shelf life and reproducibility. These considerations, coupled with the potential therapeutic benefits of pulsatile drug delivery systems, should ensure that the current high level of interest in this area would extend well in to future and result in the betterment of quality of life.
References
- O'Hagan, D.T., Jeffery, H., Roberts, M.J.J., Mc Gee, J.P. and Davis, S.S., Vaccine, 1991, 9, 768.
- O'Hagan, D.T., Rahman, D., Mc Gee, J.P., Davies, M.C., Williams, P. and Davis, S.S., Immunology, 1991, 73, 239.
- O'Hagan, D.T., Jeffery, H. and Davis, S.S., Vaccine, 1993, 11, 965.
- O'Hagan, D.T., Jeffery, H. and Davis, S.S., Int. J. Pharm., 1994, 103, 37.
- Cleland, J.L., Powell, M.F., Lim, A., Barron, L., Berman, P.W., Eastman, D.J., Nunberg, J.H., Wrin, T. and Vennari, J.C., AIDS Res. Hum. Retroviruses, 1994, 10, S21.
- Cleland, J.L., Pharm Biotechnol, 1995, 6, 439.
- Cleland, J.L., Pharm Biotechnol, 1997, 10, 1.
- Cleland, J.L., Lim, A., Barron, L., Duenas, E. and Powell, M., J. Control. Release, 1997, 47,135.
- Park. T.G., Biomaterials, 1995, 16, 1123.
- Alonso, M.J., Cohen, S., Park, T.G., Gupta, R.K., Siber, G.R. and Langer, R., Pharm. Res., 1993, 10, 945.
- Sanchez, A., Gupta, R.K., Alonso, M.J., Siber, G.R. and Langer, R., J. Pharm. Sci., 1996, 85, 547.
- Ulbrich, K., Subr, V., Podperova, P. and Buresova, M., J. Control. Release, 1995, 34, 155.
- Jiang, H.L. and Zhu, K.H., Int. J. Pharm., 2000, 194, 51
- Qui, L.Y. and Zhu, K.H., Int. J. Pharm., 2001, 219,151.
- Moriyama, K. and Yui, N., J. Control. Release, 1996, 42, 237.
- Gopferich, A., Biomaterials, 1997, 18, 397.
- Gopferich, A., Biomaterials, 1996 17, 103.
- Gopferich, A. and Langer, R., AIChE J., 1995, 41, 2292.
- Gopferich, A., J. Control. Release, 1997, 44, 271.
- Vogelhuber, W., Rotunno, P., Magni, E., Gazzaniga, A., Spruss, T., Bahardt, G., Buschaues, A., Gopferich, A., J. Control. Release, 2001, 73, 75.
- Thiel, W.J., Wyall, S.J., Barr, I. and Kleining M. In: Proc. Control. Release Society, 1994, 841.
- Cardamone, M., Lofthouse, S.A., Lucas, J.C., Lee, R.P., O'Donoghue, M. and Brandon, M.R., J. Control. Release, 1997, 47, 205.
- Kibat, P.G., Igari, Y., Wheatley, M.A., Eisen, H.N. and Langer, R., FASEB J., 1990, 4, 2533.
- Igari, Y., Kibat, P.G. and Langer, R., J. Control. Release, 1990, 14, 263.
- Ishihara, K., Kobayashi, M., Ishimura, N. and Shinohara, I., Polym. J., 1984, 16, 625.
- Obaidat, A.A. and Park, K. Biomaterials, 1997, 18, 801.
- Qui, Y. and Park, K., Adv. Drug Deliv. Rev. , 2001, 53, 321.
- Hsieh, D.S.T., Langer, R. and Folkman, J., Proc. Natl. Acad. Sci, USA., 1981, 78, 1863.
- Edelman, E.R., Kost. J., Bobeck, H. and Langer, R., J. Biomed. Mater. Res., 1985, 19, 67.
- Kost, J., Noecker, R., Kunica, E. and Langer, R., J. Biomed. Mater. Res., 1985, 19, 935.
- Saslawski, O., Weigarten, C., Beniot, J. P. and Couvereur, P., Life Sci., 1988, 1521.
- Handy, E.S., Ivkov, R., Ellis-Busby, D., Foreman, A., Braunhut, S.J., Gwost, D.U., Ardman, B. US patent No., US997863, 2006.
- Levy, D., Kost, J., Meshulam, Y. and Langer, R., J. Clin. Invest., 1989, 83, 2074.
- Kost, J., Clin. Mater., 1993, 13, 155. Back to cited text no. 34
- Machluf, M. and Kost, J., J. Biomater. Sci. Polym. Ed., 1993, 5, 147.
- Mitragotri, S., Blankschtein, D. and Langer, R., Science, 1995, 269, 850.
- Mitragotri, S., Blankschtein, D. and Langer, R., Pharm. Res., 1996, 13, 411.
- Byl, N.N., Phy. Ther., 1995, 75, 539.
- Mitragotri, S., Pharm. Res., 2000, 17, 1354.
- Kost, J., Leong, L. and Langer, R., Proc. Natl. Acad. Sci, USA., 1989, 86, 7663.
- Supersaxo, A., Kou, J.H., Teitelbaum, P. and Maskiewicz, R., J. Control. Release, 1993, 23, 157.
- Miyazaki, S., Hou, W.M. and Takada, M., Chem. Pharm. Bull., 1985, 33, 428.
- Leong, K.W., Kost, J., Mathiowitz, E. and Langer, R., Biomaterials, 1986, 7, 364.
- Kost, J., Clin. Mater., 1993, 13, 155. Back to cited text no. 44
- Okano, T., Yuim N., Yokoyama, M. and Yoshida, R., Advances in Polymerics Systems for Drug Delivery, Gordon and Breach, Yverdon, Switzerland, 1994.
- Bae, Y.H., Okano, T. and Kim, S.W., Pharm. Res., 1991, 8, 531.
- Bae, Y.H., Okano, T. and Kim, S.W., Pharm. Res., 1991, 8, 624.
- Dong, L.C. and Hoffman, A.S., J. Control. Release, 1990, 13, 21.
- Okano, T., Bae, Y. H., Jacobs, H. and Kim, S.W., J. Control. Release, 1990, 11, 255.
- Yoshida, R., Uchida, K., Kaneko, Y., Sakai, K., Kikuchi, A., Sakurai, Y. and Okano, T., Nature, 1995, 374, 240.
- Kaneko, Y., Sakai, K., Kikuchi, A., Yoshida, R., Sakurai, Y. and Okano, T., Macromolecules, 1995, 28, 7717.
- Kaneko, Y., Sakai, K., Kikuchi, A., Sakurai, Y. and Okano, T., Macromol. Chem. Macromol. Symp., 1996, 109, 41.
- Yoshida, R., Nakamura, S., Sakai, K., Aoyagi, T., Kikuchi, A., Sakurai, Y. and Okano, T., Macromolecules, 1998, 31, 6099.
- Oh, K.S., Han, S.K., Choi, Y.W., Lee, J.H., Lee, J.Y. and Yuk, S.H., Biomaterials, 2004, 25, 2393. Back to cited text no. 54
- Fisher, J.P., US patent No., US6733788, 2004.
- Mebride, J.F., Gehrker, S.H., Fisher, J.P., US patent No., US0015712, 2002.
- Kataoka, K., Haroda, A. and Nagasaki, Y., Adv. Drug. Deliv. Rev., 2001, 47, 113.
- Matsumura, Y. and Maeda, H., Cancer Res., 1986, 46, 6387.
- Chung, J.E., Yokoyama, M., Yamato, M., Aoyagi, T., Sakurai, Y. and Okano, T., J. Control. Release, 1999, 62, 115.
- Chung, J.E., Yokoyama, M. and Okano, T., J. Control. Release, 2000, 65, 93
- Kohori, F., Sakai, K., Aoyagi, T., Yokoyama, M., Sakurai, Y. and Okano, T. J. Control. Release, 1998, 55, 87.
- Kohori, F., Sakai, K., Aoyagi, T., Yokoyama, M., Sakurai, Y. and Okano, T., Colloids Surfaces B: Biointerfaces, 1999, 16, 195.
- Murdan, S., J. Control. Release, 2003, 92, 1.
- Kwon, I.C., Bae, Y.H., Okano, T., Berner, B., and Kim, S.W., Makromol. Chem. Macromol. Symp., 1990, 33, 265.
- Kwon, I.C., Bae, Y.H. and Kim, S.W., Nature, 1991, 354, 291.
- Kwon, I.C., Bae, Y.H., Okano, T.and Kim, S.W., J. Control. Release, 1991, 17, 149.
- Yuk, S.H., Cho, S.H. and Lee, H.B., Pharm. Res., 1992, 9, 955.
- Kim, S.Y. and Lee, Y.M., J. Appl. Polym. Sci., 1999, 74, 1752.
- Averitt, R.D., Westcott, S.L. and Halas, N.J., J. Opt. Soc. Amer. B., 1999, 16, 1824.
- Averitt, R.D., Sarkar, D. and Halas, N.J., Phys. Rev. Lett., 1997, 78, 4217.
- Lee, K.Y., Peters, M.C., Anderson, K.W. and Mooney, D.J., Nature, 408, 998.





