- *Corresponding Author:
- Q. H. Cheng The Second Department of Critical Care Medicine, The First Affiliated Hospital, School of Medicine, Shihezi University, Shihezi, Xinjiang Uyghur Autonomous Region 832000, P. R. China
E-mail: xunfeicheng@aliyun.com
| This article was originally published in a special issue, “Advanced Targeted Therapies in Biomedical and Pharmaceutical Sciences” |
| Indian J Pharm Sci 2023:85(1) Spl Issue “148-155” |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
Lipopolysaccharide can induce myocardial cell injury, which has been widely used in the preparation of septic cardiomyopathy models. We herein aimed to evaluate the protective effects of sodium hydrosulfide against oxidative stress and endoplasmic reticulum stress on the cell model of septic cardiomyopathy and to explore whether the protein kinase RNA-like endoplasmic reticulum kinase-eukaryotic initiation factor 2 alpha signaling pathways was involved. The sepsis induced cardiomyopathy model which was established by inducing rat cardiomyoblast cell line H9c2 cells with lipopolysaccharide and sodium hydrosulfide and endogenous hydrogen sulfide inhibitor (DL-propargylglycine) were used for intervention treatment. Then, cell viability, endogenous hydrogen sulfide levels, lactate dehydrogenase, reactive oxygen species, superoxide dismutase, glutathione and malondialdehyde were determined using commercial assay kits; enzyme-linked immunosorbent assay was used to determine creatine kinase-myoglobin binding, interleukin-6, tumor necrosis factor alpha and interleukin-10; reverse transcription-polymerase chain reaction was used to determine cystathionine gamma-lyase messenger RNA expression; and Western blot was used to determine glucose-regulated protein 78, protein kinase RNA-like endoplasmic reticulum kinase, phosphorylated protein kinase RNA-like endoplasmic reticulum kinase, eukaryotic initiation factor alpha, phosphorylated eukaryotic initiation factor alpha, activating transcription factor 4 and C/EBP-homologous protein levels. Sodium hydrosulfide increased the hydrogen sulfide contents of cells, but did not affect the messenger RNA expression of cystathionine gamma-lyase. It attenuated the decrease in cell viability caused by lipopolysaccharide; reduced the release of the myocardial enzymes like lactate dehydrogenase and creatine kinase-myoglobin binding; reduced the level of inflammatory factors like interleukin-6 and tumor necrosis factor alpha; reduced the level of oxidative stress; inhibited the expression of protein kinase RNA-like endoplasmic reticulum kinase pathway proteins. Sodium hydrosulfide treatment improves septic cardiomyopathy by inhibiting oxidative stress and endoplasmic reticulum stress via the protein kinase RNA-like endoplasmic reticulum kinaseeukaryotic initiation factor alpha pathway, which may be a theory for the treatment of septic cardiomyopathy.
Keywords
Sodium hydrosulfide, septic cardiomyopathy, endoplasmic reticulum stress, oxidative stress
Sepsis is a life-threatening uncontrolled inflammatory response to infection. The incidence of sepsis among hospitalized patients is 189 per 100 000 persons every year and it has a mortality rate of 26.7 %[1]. Sepsis-Induced Cardiomyopathy (SIC) is a reversible cardiac dysfunction caused by sepsis with a mortality rate of up to 40 %[2]. At present, SIC still lacks a unified diagnostic standard, so its reported incidences are quite different (18 %-72 %).
Commonly considered risk factors include youth, high lactate levels and a history of heart failure[3]. The specific details of sepsis pathogenesis remain unclear; however, inflammatory response disorder, mitochondrial damage, oxidative stress, coagulation disorder, immune dysfunction and Endoplasmic Reticulum Stress (ERS), are widely implicated[4]. Of these, ERS affects Unfolded Protein Response (UPR) and accumulation of misfolded proteins in the endoplasmic reticulum and when activated at mild and moderate levels, protects cells under stress conditions[5,6]. However, over or prolonged activation of UPR can cause cell dysfunction, disease and ultimately death.
UPR is mediated by the stress sensors Inositol- Requiring Enzyme 1 (IRE1), Protein Kinase Ribonucleic acid (RNA)-like Endoplasmic Reticulum Kinase (PERK) and Activating Transcription Factor 6 (ATF6) located on the endoplasmic reticulum membrane and the PERK pathway has been demonstrated to be a particularly important mediator of ERS signaling[6]. For instance, Dong et al. reported that fluorine and arsenic damage liver cells through the PERK pathway and lead to apoptosis, causing an increase of Aspartate Aminotransferase (AST) and Alanine Aminotransferase (ALT)[7] and PERK activation has been shown to promote cardiomyocyte apoptosis in ischemic cardiomyopathy[8]. Furthermore, in our previous studies, we observed elevated ERS-related protein levels in the myocardial tissue of septic mice[9].
As the third gas signal molecule identified in mammals, endogenous Hydrogen Sulfide (H2S) is producedbythreeenzymes, amongthem, Cystathionine gamma-Lyase (CSE) is highly expressed in the heart. H2S has been found to have a variety of biological effects on the cardiovascular, nervous, digestive and immune systems[10]. For example, Morpholin-4-ium 4 Methoxyphenyl (morpholino) Phosphinodithioate (GYY4137), a H2S-releasing agent, has been shown to improve the renal function of septic mice by inhibiting mitochondrial apoptosis[11] and Qiu et al. found that the reduction of H2S and the use of DL-Propargylglycine (PAG, a CSE inhibitor) increase the renal pathology score, inflammatory factors and white blood cell count in a rabbit sepsis model[12]. Furthermore, in rats with ventilator- associated pneumonia, GYY4137 reduces ERS by inhibiting the PERK pathway, reducing pulmonary edema, permeability, inflammation and apoptosis[13]. Accordingly, we reasonably hypothesized that H2S may improve septic cardiomyopathy by attenuating the PERK pathway.
Materials and Methods
Cell culture and treatment:
Rat Cardiomyoblast cell line (H9c2) cells were purchased from Procell (Wuhan, China) and cultured in a medium containing 90 % Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, United States of America (USA)), 10 % fetal bovine serum (Biological Industries (BI), Israel), and 1 % 10 000 U/ml penicillin-streptomycin (HyClone, USA) at 37° under 5 % Carbon dioxide (CO2). Escherichia coli O55: B5 Lipopolysaccharide (LPS, Sigma-Aldrich, USA) was dissolved in Double Distilled Water (DDW) at concentrations of 1 to 10 μg/ml, which were used to treat the cells for 12 h to 48 h.
Stock solutions of Sodium Hydrosulfide (NaHS) (>98 %, Sigma-Aldrich, USA) and PAG (Sigma- Aldrich USA) were prepared in DDW. The cells were treated with NaHS at 20 to 150 mM for 0.5 h prior to LPS and/or PAG stimulation. The control group was administered with the same amount of DDW. The H9c2 cells were divided into 7 groups: Control group, LPS group, NaHS group, LPS+NaHS group, PAG group, LPS+PAG group and LPS+NaHS+PAG group. According to the results shown in fig. 1 and fig. 2 along with existing research, we used LPS at 5 µg/ml, 24 h, NaHS at 50 µM and PAG at 2 mM for all subsequent experiments[14-16].
Cell viability:
Cell Counting Kit-8 (CCK-8) (Dojindo Japan) kit was used to test cell viability. Briefly, H9c2 cells (5×103 cells/well) were seeded in 96-well plates overnight and then treated with LPS and/or NaHS and/or PAG. At the end of the specified treatment, add 10 µl of CCK-8 solution to each well and then the plate was incubated for another 2 h at 37°. A microplate reader (Thermo Fisher, USA) was then used to measure absorbance at 450 nm. Cell viability (%) was calculated as (Atreatment-Ablank)/(Acontrol-Ablank)×100 %, where A=Absorbance.
Determination of Lactate Dehydrogenase (LDH):
H9c2 cells were seeded in six-well plates (2×105 cells/well) for 24 h and then treated with LPS and/ or NaHS and/or PAG. After the specified treatment, the supernatant was subjected to LDH determination using commercial kits (Nanjing Institute of Bioengineering Nanjing, China) according to the operation manuals.
Determination of H2S, Glutathione (GSH), Malondialdehyde (MDA) and Superoxide Dismutase (SOD):
H9c2 cells were seeded in a six-well plates (2×105 cells/well) for 24 h and then treated with LPS or/and NaHS or/and PAG. After the specified treatment, we collected the cells and determined H2S levels using a commercial kit (Nanjing Institute of Bioengineering Nanjing, China) according to the operation manual.
Determination of Tumor Necrosis Factor alpha (TNF-α), Interleukin-6 (IL-6), Interleukin-10 (IL-10) and Creatine Kinase-Myoglobin Binding (CK-MB):
After completing the aforementioned treatment, the Enzyme-Linked Immunosorbent Assay (ELISA) kits, EK-00526, EK-0414, EK-0418, (Boster, China) and bs-14049R-2 (Shanghai China) were used to determine TNF-α, IL-6, IL-10 and CK-MB levels, respectively, in the supernatant medium according to the manufacturer’s instructions.
Determination of CSE messenger RNA (mRNA):
After the experimental treatments, the cells were washed with pre-cooled PBS and the total RNA was extracted with TRIzol (Life, USA). A RevertAid First Strand complementary Deoxyribonucleic Acid (cDNA) synthesis kit (Thermo Scientific, USA) was used to transcribe the extracted RNA into cDNA.
The primer sequences were as follows: CSE, Forward: 5’-GGCCTGGTGTCTGTTAATTGT-3’, Reverse: 5’-GCCATTCCGTTTTTGAAATGCT-3’, Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH), Forward: 5’-CGCTCTCTGCTCCTCC TGTTC-3’, Reverse: 5’-ATCCGTTGACTCCGACC TTCAC-3’. Reverse Transcription-Polymerase Chain Reaction (RT-PCR) was performed using an SYBR Green PCR kit (QuantiNova, Germany) and a 7500 Fast RT-PCR system (Life, USA).
Determination of Reactive Oxygen Species (ROS):
After completing the aforementioned treatment, the cells were washed twice with DMEM and inoculate with 5 µmol/l Dichloro-Dihydro-Fluorescein Diacetate (DCFH-DA) (Solarbi, China) for 15 min at 37°. Wash again for 3 times and observe under a fluorescence microscope at 488 nm.
Protein determination:
Cell protein was extracted with Radioimmunoprecipitation Assay (RIPA) lysis buffer (Solarbio, China). The quality of the whole cell extract was quantified with NanoDropTM 2000 (Thermo Scientific, USA). Part of the protein PERK and phosphorylated PERK (pPERK) were electrophoresed in 8 % Sodium Dodecyl Sulphate- Polyacrylamide Gel (SDS-PAG) and the remaining proteins were electrophoresed in 10 % SDS-PAG. Then they were transferred to the Polyvinylidene Fluoride (PVDF) membrane, where PERK and pPERK were at 80 V, 2 h and the rest of the proteins were at 80 V for 1 h. The membrane was blocked with Bovine Serum Albumin (BSA) at room temperature for 1 h or 3 h and incubated overnight at 4° with primary antibody. Specific antibodies for Glucose Regulated Protein 78 (GRP78) (ab-21685; dilution 1:700), PERK (ab-79483; dilution 1:500), pPERK (ab-192591, dilution 1:500), eukaryotic Initiation Factor alpha (eIFα) (ab-28726; dilution 1:500), phosphorylated eIFα (peIFα) (ab-227593, dilution 1:500), Activating Transcription Factor 4 (ATF4) (ab-85049; dilution 1:500) and CCAAT/Enhancer- Binding Protein-Homologous Protein (CHOP) (ab- 11419; dilution 1:1000) were purchased from Abcam. Beta (β)-actin antibody (TA-09; dilution 1:1000) was purchased from Beijing Biotechnology (Beijing, China). Following incubation with goat anti-rabbit (ZB 2301; dilution 1:10 000) and goat anti-mouse (ZB 2305; dilution 1:10 000) Immunoglobulin G (IgG) secondary antibodies (Beijing Biotechnology, China) at room temperature for 1 h, the positive signals were developed with a Super Electrochemiluminescence (ECL) kit (Millipore, USA) according to the manufacturer’s instructions. The intensity of each band was quantified using Image Lab Software (Thermo Scientific, USA).
Statistical analysis:
All represented data are displayed as the mean±Standard Deviation (SD). Using the Statistical Package for the Social Sciences (SPSS) version 22.0 (IBM, USA), one-way Analysis of Variance (ANOVA) was utilized to analyze the data. Statistical difference was achieved when p values are less than 0.05. All experiments are repeated 3 times.
Results and Discussion
Effect of LPS on H9c2 cells is shown in fig. 1. As shown in fig. 1, compared with the control group, the viability of the H9c2 cells significantly decreases with increasing LPS stimulation concentration (p<0.05) and at the same concentration, decreases with increasing treatment time. LPS also stimulates the release of LDH in cells, which also increases with increasing treatment concentration and time. Considering that the average activity of 5 μg/ml LPS is 54.44 %, the average release of LDH is 197 U/l.

Fig. 1: LPS-induced H9c2 cell damage
Note: (A) H9c2 cell viabilities for different concentrations and treatment times,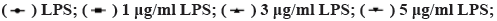
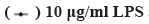 and (B) Contents of LDH in the supernatant of H9c2 cells at different concentrations and times,
and (B) Contents of LDH in the supernatant of H9c2 cells at different concentrations and times,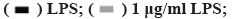
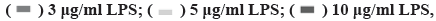 *p<0.05 compared with the control group at the same time-point and #p<0.05 compared with 12 h at the same concentration
*p<0.05 compared with the control group at the same time-point and #p<0.05 compared with 12 h at the same concentration
Thus, the treatment with LPS at 5 μg/ml for 24 h establishes a viable sepsis model.
Effect of NaHS on LPS-induced cell damage is shown in fig. 2. As shown in fig. 2A, exogenous H2S increases the content of H2S in H9c2 cells (p<0.05), but its content will decrease with time, there is no significant difference between the LPS group and the NaHS group at 48 h (p>0.05). Furthermore, as shown in fig. 2B and fig. 2C, NaHS can improve the decrease in cell viability caused by LPS and reduce the release of LDH caused by LPS. However, its effect is not concentration dependent. Therefore, it was necessary to choose an appropriate concentration for further study. At NaHS concentrations above 100 µM, no further increase in cell viability or reduction in LDH release is observed. In fact, negative effects are observed at 150 µM NaHS. At 50 μM, the treatment effect is optimal, so this concentration was used for further experiments.

Fig. 2: NaHS ameliorates cell damage induced by LPS
Note: (A) H2S contents in H9c2 cells; (B) Differences in cell viability between groups and (C) Differences in LDH between groups,
 *p<0.05 compared with the control group at the same time-point and #p<0.05 compared with 12 h at the same concentration
*p<0.05 compared with the control group at the same time-point and #p<0.05 compared with 12 h at the same concentration
Changes in endogenous H2S and inflammatory factors levels after administration of NaHS or PAG was shown in fig. 3. As shown in fig. 3A, compared with that of the control group, the expression of CSE mRNA for the LPS and PAG groups is significantly decreased (p<0.05). However, CSE mRNA expression for the LPS and LPS+NaHS groups are not significantly different (p>0.05). As shown in fig. 3B, compared with that of the control group, the H2S levels in the LPS and PAG groups are significantly decreased (p<0.05). However, it is significantly increased for the NaHS group compared with the LPS group. Furthermore, it is significantly increased in the LPS+NaHS group (p<0.05) and significantly increased for the PAG+NaHS group (p<0.05). As shown in fig. 3C-fig. 3E, compared with the control group, cell viability decreases and CK-MB and LDH are significantly increased for the LPS group (p<0.05). Compared with the LPS group, NaHS can reduce LDH and CK-MB levels and increase cell viability (p<0.05); compared with the LPS group, the levels of LDH increased (p<0.05) and the cell viability decreased (p<0.05) after PAG administration. As shown in fig. 3F-fig. 3H, compared with the control group, LPS increases the release of the inflammatory factors IL-6, TNF-α and IL-10 (p<0.05). After administration of NaHS, pro-inflammatory IL-6 and TNF-α are significantly reduced, anti-inflammatory IL-10 increased compared with those of the LPS group (p<0.05). After PAG administration, pro- inflammatory IL-6, TNF-α is increased (p<0.05) and anti-inflammatory IL-10 is decreased.

Fig. 3: Effects of administration of NaHS or PAG
Note: (A) CSE mRNA expression in each group; (B) H2S contents for each group; (C) Cell viabilities for each group; (D) LDH levels for each group; (E) CK-MB levels for each group; (F) IL-6 levels for each group; (G) TNF-α levels for each group and (H) IL-10 levels for each group, *p<0.05 compared with the control group and #p<0.05 compared with the LPS group
Changes in oxidative stress after administration of NaHS or PAG are shown in fig. 4. As shown in fig. 4A-fig. 4D, compared with the control group, LPS increased the release of ROS and MDA and inhibited the release of SOD and GSH (p<0.05). After administration of NaHS, ROS and MDA are significantly reduced, GSH and SOD increased compared with those of the LPS group (p<0.05). After PAG administration, ROS and MDA are increased (p<0.05), GSH and SOD is decreased.

Fig. 4: Effects of administration of NaHS or PAG on oxidative stress
Note: (A) ROS expression in each group (magnification, 200×) and (B-D) MDA, GSH, SOD contents for each group, *p<0.05 compared with the control group and #p<0.05 compared with the LPS group
ERS and changes in PERK-pathway-related proteins after administration of NaHS or PAG are shown in fig. 5. As shown in fig. 5, compared with the control group, GRP78, PERK, pPERK, eIFα, peIFα, ATF4 and CHOP are significantly increased in the LPS group (p<0.05). After administration of NaHS, these proteins are reduced compared with the LPS group. After administration of PAG, the above-mentioned proteins are significantly increased (p<0.05).

Fig. 5: Shows the different expressions of ERS-related proteins in cardiomyocytes after NaHS or PAG
Note: *p<0.05 compared with the control group and #p<0.05 compared with the LPS group
As an endogenous gas molecule, H2S has been shown to play a role in many biological processes, including those related to inflammatory conditions. Specifically, H2S can improve inflammation in cardiovascular diseases[17]. NaHS is a quick release and relatively stable reagent of H2S, which is often used as the donor of exogenous H2S. Therefore, we used NaHS to increase H2S content in both in vitro models. The results showed that NaHS increased H2S content and cell viability, reduced the release of inflammatory factors, inhibited the release of oxygen free radicals and alleviated the myocardial injury induced by sepsis.
NaHS is a fast releasing H2S agent, but it does not affect the synthesis of endogenous H2S. Furthermore, CSE mRNA expression is not affected by NaHS. Therefore, as shown in fig. 2, the H2S content in the 12 h treatment group is increased compared with the control group, but no significant difference between the groups at 48 h is observed. H2S seems to play a complex role in sepsis and its effect may be related to dose and species. As shown in fig. 2, NaHS does not improve cell viability in a concentration dependent manner, which is similar to the findings of Bernardini et al.[18]. It was shown that high concentration of NaHS (300 µM) could not further improve the vitality of porcine vascular mesenchymal stem cells and even stagnated the cell cycle at high concentration of NaHS.
The pathogenesis of sepsis is still not completely clear, but it is clear that one of its main mechanisms is the imbalance of inflammatory response in the body and the “inflammatory storm” caused by the release of a large number of inflammatory factors in a short period of time and many studies have shown that an increase of inflammatory factors is related to poor prognosis[3,19]. The CHOP and the ATF4 are added into the metabolism and continuous activation of ATF4 not only induces apoptosis or necrosis of cells, but also activates Nuclear Factor kappa B (NF- κB) through toll-like receptor activation, resulting in the release of a large number of inflammatory factors. By measuring the expression of CHOP and ATF4 proteins and the release of representative inflammatory factors (IL-6, IL-10 and TNF-α), the mechanism can be clarified. Clinical trials have shown that using ulinastatin to improve sepsis leads to a significant decrease in the inflammatory factors IL-6 and TNF-α, while the anti-inflammatory factor IL-10 is increased[20]. This is consistent with our research, which shows that NaHS can ameliorate LPS-induced cell damage.
The activation of PERK pathway in ERS promotes the transcription of CHOP gene and the activation of CHOP directly causes the release of ROS. At the same time, ERS causes calcium ion metabolism disorder, which damages mitochondrial respiratory chain and also directly causes ROS release[21]. When ERS occurs, the accumulation of misfolded proteins leads to increased ROS release and consumption of antioxidant enzymes in which reduced GSH reduces protein misfolding. Secondly, some active oxygen components can affect the protein folding in the endoplasmic reticulum and induce internal reticulum stress. These two causes and affects each other and promote each other to cause the release of oxygen free radicals in a short period of time. It has been found that the myocardial damage caused by sepsis can be improved by inhibiting inflammation and oxidative stress[22]. In our study, ROS-a marker of oxygen free radical release, MDA-a metabolite of lipid oxidation reaction and GSH and SOD-which represent the antioxidant enzyme system, were measured. Studies showed that in LPS group, intracellular CHOP, ROS and MDA increased, GSH and SOD expression decreased, accompanied by decreased cell viability and a large amount of myocardial enzyme release.
The results showed that LPS stimulated H9c2 cardiomyocytes, induced a large number of ROS, lipid peroxides production, consumption of antioxidant enzymes, resulting in the accumulation of oxygen free radicals in the cells, resulting in cell damage. It was indicated that exogenous NaHS could alleviate mitochondrial damage, inhibit the generation of oxygen free radicals, antagonize the ROS reaction and improve the myocardial damage caused by sepsis.
ERS is an important response of cell to maintain physiological function, but the excessive activation of ERS will lead to cell death and then lead to tissue damage and organ failure. As it is known to all, abnormal increase of ERS-related proteins can be observed in various pathological states and in the previous study of our research group, abnormal increase of marker proteins of ERS (GRP78, CHOP) can be observed in the myocardial tissue of sepsis mice[23]. In our in vitro experiments, we also observed the increase of GRP78, a marker of ERS, in H9c2 cardiomyocytes in the LPS group. To further clarify the mechanism, we examined PERK, one of the ERS receptors located in the endoplasmic reticulum lumen and its downstream related proteins.
During ERS, the Binding immunoglobulin Protein (BiP) is released by PERK, which itself is activated by phosphorylation. Phosphorylated PERK activates eukaryotic Initiation Factor 2 alpha (eIF2α) to inhibit mRNA transcription, thereby reducing protein synthesis and activating ATF4. Sustained activation of the PERK-eIF2α pathway leads to the activation of CHOP, which induces apoptosis or autophagy[24]. In this study, it was confirmed that GRP78, pPERK, peIF2α, ATF4 and CHOP proteins were significantly increased in LPS group compared with control group. These results indicated that LPS damage to cardiomyocytes activated the PERK pathway in the UPR, resulting in blocked protein synthesis and folding in the endoplasmic reticulum, and accumulation of misfolded and unfolded proteins in the endoplasmic reticulum, leading to ERS. In LPS+NaHS group and LPS+PAG+NaHS group, the expression of PERK pathway protein was significantly decreased. The expression of PAG was further upregulated in LPS group. Combined with the above results, it was suggested that NaHS increased the content of H2S in cells, effectively inhibited the activation of the PERK pathway in ERS, reduced the release of myocardial enzymes and improved cell viability. Therefore, sepsis is thought to activate the PERK pathway in ERS and exogenous H2S can inhibit the activation of PERK pathway related proteins to alleviate sepsis induced myocardial damage.
To sum up, our research proves that the protective effect of NaHS on the cell model of septic cardiomyopathy is done at least partially through PERK-eIF2α signaling pathway, inhibiting oxidative stress and ERS, thereby inhibiting the pathological progress of septic cardiomyopathy. Our research is still limited to cell experiments and further verification of animal models and even clinical practice is needed in future research. Although the specific molecular mechanism of NaHS against septic cardiomyopathy has not been fully determined, the current work may provide new insights into the potential clinical application of NaHS as a drug for treating septic cardiomyopathy.
Funding:
This study was supported by the National Natural Science Foundation of China (No. 81860336).
Author’s contributions:
Xin Peng and Guodong Cao contributed equally to this work.
Conflict of interests:
The authors have no potential conflicts of interest to disclose.
References
- Fleischmann-Struzek C, Mellhammar L, Rose N, Cassini A, Rudd KE, Schlattmann P, et al. Incidence and mortality of hospital-and ICU-treated sepsis: Results from an updated and expanded systematic review and meta-analysis. Intensive Care Med 2020;46:1552-62.
[Crossref] [Google Scholar] [PubMed]
- Liu YC, Yu MM, Shou ST, Chai YF. Sepsis-induced cardiomyopathy: Mechanisms and treatments. Front Immunol 2017;8:1021.
[Crossref] [Google Scholar] [PubMed]
- Beesley SJ, Weber G, Sarge T, Nikravan S, Grissom CK, Lanspa MJ, et al. Septic cardiomyopathy. Crit Care Med 2018;46(4):625-34.
[Crossref] [Google Scholar] [PubMed]
- Huang M, Cai S, Su J. The pathogenesis of sepsis and potential therapeutic targets. Int J Mol Sci 2019;20(21):1-31.
[Crossref] [Google Scholar] [PubMed]
- Hetz C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 2012;13(2):89-102.
[Crossref] [Google Scholar] [PubMed]
- Pluquet O, Pourtier A, Abbadie C. The unfolded protein response and cellular senescence. A review in the theme: Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am J Physiol Cell Physiol 2015;308(6):C415-25.
[Crossref] [Google Scholar] [PubMed]
- Dong N, Feng J, Xie J, Tian X, Li M, Liu P, et al. Co-exposure to arsenic-fluoride results in endoplasmic reticulum stress-induced apoptosis through the PERK signaling pathway in the liver of offspring rats. Biol Trace Elem Res 2020;197:192-201.
[Crossref] [Google Scholar] [PubMed]
- Guo XF, Yang XJ. Endoplasmic reticulum stress response in spontaneously hypertensive rats is affected by myocardial ischemia reperfusion injury. Exp Ther Med 2015;9(2):319-26.
[Crossref] [Google Scholar] [PubMed]
- Li L, Peng X, Guo L, Zhao Y, Cheng Q. Sepsis causes heart injury through endoplasmic reticulum stress-mediated apoptosis signaling pathway. Int J Clin Exp Pathol 2020;13(5):964-71.
[Google Scholar] [PubMed]
- Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J 2001;20(21):6008-16.
[Crossref] [Google Scholar] [PubMed]
- Wang C, Du J, Du S, Liu Y, Li D, Zhu X, et al. Endogenous H2S resists mitochondria-mediated apoptosis in the adrenal glands via ATP5A1 S-sulfhydration in male mice. Mol Cell Endocrinol 2018;474:65-73.
[Crossref] [Google Scholar] [PubMed]
- Qiu H, Chen X, Luo Z, Zhao L, Zhang T, Yang N, et al. Inhibition of endogenous hydrogen sulfide production exacerbates the inflammatory response during urine?derived sepsis?induced kidney injury. Exp Ther Med 2018;16(4):2851-8.
[Crossref] [Google Scholar] [PubMed]
- Ge X, Sun J, Fei A, Gao C, Pan S, Wu Z. Hydrogen sulfide treatment alleviated ventilator-induced lung injury through regulation of autophagy and endoplasmic reticulum stress. Int J Biol Sci 2019;15(13):2872-84.
[Crossref] [Google Scholar] [PubMed]
- Zhang X, Zhao Y, Bai D, Yuan X, Cong S. Schizandrin protects H9c2 cells against lipopolysaccharide?induced injury by downregulating Smad3. J Biochem Mol Toxicol 2019;33(5):e22301.
[Crossref] [Google Scholar] [PubMed]
- Yang H, Mao Y, Tan B, Luo S, Zhu Y. The protective effects of endogenous hydrogen sulfide modulator, S-propargyl-cysteine, on high glucose-induced apoptosis in cardiomyocytes: A novel mechanism mediated by the activation of Nrf2. Eur J Pharmacol 2015;761:135-43.
[Crossref] [Google Scholar] [PubMed]
- Pan LL, Liu XH, Gong QH, Zhu YZ. S-Propargyl-cysteine (SPRC) attenuated lipopolysaccharide-induced inflammatory response in H9c2 cells involved in a hydrogen sulfide-dependent mechanism. Amino Acids 2011;41:205-15.
[Crossref] [Google Scholar] [PubMed]
- Wallace JL, Vaughan D, Dicay M, MacNaughton WK, de Nucci G. Hydrogen sulfide-releasing therapeutics: Translation to the clinic. Antioxid Redox Signal 2018;28(16):1533-40.
[Crossref] [Google Scholar] [PubMed]
- Bernardini C, La Mantia D, Nesci S, Salaroli R, Algieri C, Pagliarani A, et al. Effects of hydrogen sulfide donor NaHS on porcine vascular wall-mesenchymal stem cells. Int J Mol Sci 2020;21(15):5267:1-12.
[Crossref] [Google Scholar] [PubMed]
- Ma X, Chang W, Zhang C, Zhou X, Yu F. Staphylococcal Panton-Valentine leukocidin induces pro-inflammatory cytokine production and nuclear factor-kappa B activation in neutrophils. PLoS One 2012;7(4):e34970.
[Crossref] [Google Scholar] [PubMed]
- Wang H, Liu B, Tang Y, Chang P, Yao L, Huang B, et al. Improvement of sepsis prognosis by ulinastatin: A systematic review and meta-analysis of randomized controlled trials. Front Pharmacol 2019;10:1-11.
[Crossref] [Google Scholar] [PubMed]
- Zhang B, Liu Y, Zhang JS, Zhang XH, Chen WJ, Yin XH, et al. Cortistatin protects myocardium from endoplasmic reticulum stress induced apoptosis during sepsis. Mol Cell Endocrinol 2015;406:40-8.
[Crossref] [Google Scholar] [PubMed]
- Chen X, Wang Y, Xie X, Chen H, Zhu Q, Ge Z, et al. Heme oxygenase-1 reduces sepsis-induced endoplasmic reticulum stress and acute lung injury. Mediators Inflamm 2018:1-10.
[Crossref] [Google Scholar] [PubMed]
- Liu W, Liu K, Zhang S, Shan L, Tang J. Tetramethylpyrazine showed therapeutic effects on sepsis-induced acute lung injury in rats by inhibiting endoplasmic reticulum stress protein kinase RNA-like endoplasmic reticulum kinase (PERK) signaling-induced apoptosis of pulmonary microvascular endothelial cells. Med Sci Monit 2018;24:1225-31.
[Crossref] [Google Scholar] [PubMed]
- Sun W, Yang J, Zhang Y, Xi Y, Wen X, Yuan D, et al. Exogenous H2S restores ischemic post-conditioning-induced cardioprotection through inhibiting endoplasmic reticulum stress in the aged cardiomyocytes. Cell Biosci 2017;7(1):1-4.
[Crossref] [Google Scholar] [PubMed]





