- Corresponding Author:
- D. J. S. Sharmila
Department of Bioinformatics, Karunya University, Karunya Nagar, Coimbatore-641 114
E-mail: djssharmila@gmail.com
| Date of Submission | 26 October 2009 |
| Date of Revision | 10 June 2010 |
| Date of Acceptance | 26 July 2010 |
| Indian J Pharm Sci, 2010, 72 (4): 449-457 |
Abstract
Synthetic sialic acid analogues with multiple modifications at different positions(C-1/C-2/C-4/C-8/C-9) are investigated by molecular mechanics and molecular dynamics to determine their conformational preferences and structural stability to interact with their natural receptors. Sialic acids with multiple modifications are soaked in a periodic box of water as solvent. Molecular mechanics and a 2 nanosecond molecular dynamics are done using amber force fields with 30 picosecond equilibrium. Direct and water mediated hydrogen bonds existing in the sialic acid analogues, aiding for their structural stabilization are identified in this study. The accessible conformations of side chain linkages of sialic acid analogues holding multiple substituents are determined from molecular dynamics trajectory at every 1ps interval. Transitions between different minimum energy regions in conformational maps are also noticed in C-1, C-2, C-4, C-8 and C-9 substituents. Docking studies were done to find the binding mode of the sialic acid analogues with Influenza hemagglutinin. This finding provides stereo chemical explanation and conformational preference of sialic acid analogues which may be crucial for the design of sialic acid analogues as inhibitors for different sialic acid specific pathogenic proteins such as influenza toxins and neuraminidases.
Keywords
AMBER, molecular dynamics, molecular mechanics, molecular modeling, sialic acid analogues
The binding of a virus particle to the surface receptors of a host cell is mediated by viral proteins, which specifically recognize receptor determinants, such as peptides, lipids or carbohydrates[1]. The receptor determinants of Infl uenza virus are shown to be sialic acid residues, which constitute more than 20 naturally occurring derivatives of neuraminic acid collectively referred to as a family of sialic acids. These are found widely distributed in animal tissues and in bacteria, especially in glycoproteins and gangliosides. Influenza A and B bind to the most abundant sialic acid, N-acetyl neuraminic acid (Neu5Ac or NANA). The binding of infl uenza A virus is mediated by the major virus surface glycoprotein, hemagglutinin (HA) that binds to the terminal sialic acid residues as the fi rst step of viral infection and mediates both the initial attachment of virus to target cells and the subsequent fusion of the viral and cell membranes[2]. The region of the HA involved in receptor binding has been deduced from studies of mutant HAs with different binding specificities to involve a pocket of amino acids at its membrane distal surface[3]. The three-dimensional structure of infl uenza virus HAs complexed with cell receptor analogues show neuraminic acid bound to this pocket fundamentally through hydrogen bonds and van der Waals interactions[4].
Based on this knowledge, it should, in principle be possible to fi nd a neuraminic acid analogue that mimics the cell receptor and thus preferentially binds to the virus, thereby blocking attachment. One approach in the design of high-affi nity inhibitors is to use Neu5Ac (or its 2α-O-methyl derivative) as a scaffold and to modify its functional groups in order to increase its affi nity for the HA. The confi guration of neuraminic acid places the carboxylate in the axial position is the alpha-anomer of neuraminic acid.
Present work was initiated with the modeling of the alpha-anomer of neuraminic acid and its derivatives having multiple substitutions at C-1, C-2, C-4, C-8 and C-9 positions. Molecular mechanics and molecular dynamics calculations were performed. The conformational behaviour of varying substituent holding side chains of neuraminic acid in aqueous environment were studied. The direct and water-mediated hydrogen bonds, which played a major role in the structural stability of neuraminic acid were also analyzed. Docking studies were done to study the binding mode of neuraminic acid derivatives into the binding pocket of Infl uenza HA.
Materials and Methods
The modeled neuraminic acid derivatives with multiple substituents are shown in Table 1. The neuraminic acid derivatives are designed as described by Sauter, et al., 1992[5] and Bianco, et al., 1998[6]. The initial conformations of the different substituent holding side chains of neuraminic acid analogues are defined as follows: for C-1 substitution, γ1=0 when C3-C2 cis to C1-O1A; γ2=0 when C2-C1 cis to O1A-C11, where C11 is the carbonyl carbon atom from the substitution group. For C-2 substitution, θ1=0 when C4-C3 cis to C2-O2; θ2=0 when C3-C2 cis to O2-C12, where C12 is the carbon attached to the substituent group. For C-4 substitution, β1=0 when C2-C3 cis to C4-O4; β2=0 when C3-C4 cis to O4-C13, where C13 is the carbon atom attached to the substituent group. For C-8 substitution, χ1=0 when C5-C6 cis to C7-C8; χ3=0 when C6-C7 cis to C8-O8. For C-9 substitution, χ1=0 when C5-C6 cis to C7-C8; χ2=0 when C6-C7 cis to C8-C9.
| Sialic acid derivative | Substituents | ||||
|---|---|---|---|---|---|
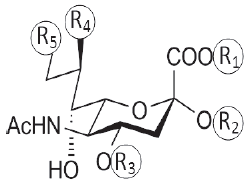 |
R1 | R2 | R3 | R4 | R5 |
| N-acetyl Neuraminic Acid methyl | H | H | H | OH | OH |
| 5-N-acetyl neuraminate | CH3 | H | H | OH | OH |
| methyl 2α-O-methyl-5-N-acetyl neuraminate | CH3 | CH3 | H | OH | OH |
| benzyl 2α-O-methyl-5-N-acetyl-8,9-O-isopropylidene neuraminate | CH2Ph | CH3 | H | OCH3 | OCH3 |
| benzyl 2α-O-methyl-4-O-capriloyl-5-N-acetyl-8,9-O-isopropylidene neuraminate | CH2Ph | CH3 | CO2(CH2)6CH3 | OCH3 | OCH3 |
| benzyl 2α-O-methyl-4-O-capryloil-5-N-acetyl neuraminate | CH2Ph | CH3 | CO2(CH2)6CH3 | OH | OH |
| 2α-O-methyl-4-O-capriloyl-5-N-acetyl neuraminic acid | H | CH3 | CO2(CH2)6CH3 | OH | OH |
| benzyl-2α-O-methyl-4-O-(8-morpholin)-capriloyl-5-N-acetyl-8,9-O-isopropylidene-neuraminate | CH2Ph | CH3 | CO2(CH2)7O(CH2CH2)2NH | OCH3 | OCH3 |
| benzyl-2α-O-methyl-4-O-(8-morpholin)-capriyloyl-5-N-acetyl-neuraminate | CH2Ph | CH3 | CO2(CH2)7O(CH2CH2)2NH | OH | OH |
| 2α-O-methyl-4-O-(8-morpholin)-capriloyl-5-N-acetyl neuraminic acid | H | CH3 | CO2(CH2)7O(CH2CH2)2NH | OH | OH |
| 5-N-acetyl-9-amino-9-deoxy neuraminic acid | H | H | H | OH | NH2 |
Table 1: Neuraminic acid analogues with multiple modifications
Molecular mechanics calculations were carried out in the Pentium IV workstation using SANDER module of software AMBER10[7]. The force fields AMBER ff03 and gaff (general amber force fi eld) were used. Water molecules are added from the solvent library of AMBER10 and care is given to maintain the number of water molecules to be same for all the neuraminic acid derivatives.
A periodic box enclosing the neuraminic acid analogues in solution is constructed to turn it into a periodic system for the simulation programs and periodic boundary conditions are applied on constant volume (Table 2). The non bonded pair list was updated for every 10 steps. The non-bonded cutoff was specifi ed as 8Å. For initial 100 cycles steepest descent method was used, then conjugate gradient is switched on. The convergence criterion for the energy gradient is less than 0.01kcal mole-1. The energy minimized structures for all the neuraminic acid derivatives are intensively analyzed through graphics software VEGA ZZ to work out the direct and solvent mediated hydrogen bonds.
| Sialic acid derivative | No of atoms | Box size(Å3) |
|---|---|---|
| N-acetyl Neuraminic Acid | 40 | 25.071×22.641×23.266 |
| methyl 5-N-acetyl | 43 | 23.338×22.854×24.077 |
| neuraminate | ||
| methyl 2α-O-methyl-5-N- | 46 | 24.734×23.343×22.600 |
| acetyl neuraminate | ||
| benzyl 2α-O-methyl-5- | 62 | 24.734×23.343×24.077 |
| N-acetyl-8,9-O- | ||
| isopropylidene | ||
| neuraminate | ||
| benzyl 2α-O-methyl-4- | 85 | 21.402×30.414×21.022 |
| O-capriloyl-5-N-acetyl- | ||
| 8,9-O-isopropylidene | ||
| neuraminate | ||
| benzyl 2α-O-methyl-4- | 79 | 21.402×29.461×21.362 |
| O-capryloil-5-N-acetyl | ||
| neuraminate | ||
| 2α-O-methyl-4-O- | 66 | 23.338×21.568×26.582 |
| capriloyl-5-N-acetyl | ||
| neuraminic acid | ||
| benzyl-2α-O-methyl- | 98 | 20.772×32.158×21.362 |
| 4-O-(8-morpholin)- | ||
| capriloyl-5-N-acetyl- | ||
| 8,9-O-isopropylidene- | ||
| neuraminate | ||
| benzyl-2α-O-methyl-4-O- | 92 | 20.086×30.511×23.266 |
| (8-morpholin)-capriyloyl- | ||
| 5-N-acetyl-neuraminate | ||
| 2α-O-methyl-4-O-(8- | 79 | 25.071×20.106×29.273 |
| morpholin)-capriloyl-5-N- | ||
| acetyl neuraminic acid | ||
| 5-N-acetyl-9-amino-9- | 41 | 25.002×20.983×24.417 |
| deoxyneuraminic acid |
Table 2: The box size and number of atoms in each neuraminic acid analogue with multiple modifications.
To understand the conformational dynamics of the neuraminic acid analogues in aqueous environment, molecular dynamics calculations were performed over a period of 30ps equilibration followed by a 2 ns production run with explicit inclusion of water molecules. The width of integration step of the MD simulation was 1fs. The history of information was recorded for every 1000 steps of trajectory which resulted in 2000 structures. The temperature was maintained to be 300K. The total simulation time was around 17 h for each molecule. The MD trajectory information collected for every 1ps were analyzed using PTRAJ (Trajectory Analysis) module of AMBER10 package.
Docking studies were done for all the 18 sialic acid analogues using Schrodinger (maestro). The protein is prepared by optimizing and minimizing the structure using Protein Preparation Wizard. The grid is generated using Receptor Grid Generation by picking the reference ligand which is already present in the PDB structure. HTVS is performed by importing 18 minimized sialic acid analogues using GLIDE module of Schrodinger software[8,9]. Induced fit is carried out for the five top scoring sialic acid analogues using Schrodinger to predict the ligand-induced conformational changes in receptor active sites[10].
Results
Relative energy of neuraminic acid derivatives
The relative energy is calculated for all the 10 neuraminic acid derivatives with respect to the absolute minimum energy of neuraminic acid (-2395.1 kcal/mol). The minimum energy conformations of neuraminic acid derivatives with respect to their relative energy are displayed in Table 3. Analogue 10 (5-N-acetyl-9-amino-9-deoxy neuraminic acid) is found to have the minimum energy equal to the minimum energy of neuraminic acid. Analogue 6 (2α-O-methyl-4- O-capriloyl-5-N-acetyl neuraminic acid) has the relative minimum energy of 14.7 kcal/mol.
It is noted from Table 3 that, in the minimum energy conformation for Analogue 10 (5-N-acetyl-9-amino-9- deoxy neuraminic acid), the side chain conformations (χ1, χ2) prefer values around (-177.11°,-90.33°). For Analogue 6 (2α-O-methyl-4-O-capriloyl-5-N-acetyl neuraminic acid), the substituent dwelling side chain torsion angles (θ1,θ2) and (β1, β2) prefer the values (-69.48°,167.13°) and (-170.66°,-71.12°), respectively.
| Neuraminic acid derivative No | Relative energy kcal/mol | C1 substitution (γ1, γ2) (deg.) | C2 substitution (θ1,θ2) (deg.) | C4 substitution (β1, β2) (deg.) | C8 substitution (χ1, χ3) (deg.) | C9 substitution (χ1, χ2) (deg.) |
|---|---|---|---|---|---|---|
| N-acetyl | 0 | - | - | - | - | - |
| Neuraminic Acid | ||||||
| 1 | 49.1 | (-52.53,-179.14) | - | - | - | - |
| 2 | 73.4 | (-70.20,171.27) | (-74.43,171.39) | - | - | - |
| 3 | 35.6 | (-100.06,179.65) | (-68.74,-178.68) | - | (175.31,49.82) | (175.31,-73.05) |
| 4 | 76.7 | (-73.32,177.19) | (-72.78,172.96) | (-169.32,-69.33) | (164.29,58.31) | (164.29,-65.13) |
| 5 | 28.1 | (-76.46,174.20) | (-63.98,174.14) | (-175.96,-73.03) | - | - |
| 6 | 14.7 | - | (-69.48,167.13) | (-170.66,-71.12) | - | - |
| 7 | 115.9 | (-68.53,178.43) | (-62.98,172.91) | (-177.16,-72.77) | (-175.04,47.84) | (-175.04,-75.38) |
| 8 | 60.8 | (-93.66,-174.97) | (-63.28,172.42) | (-174.64,-72.59) | - | - |
| 9 | 75.4 | - | (-72.59,175.08) | (-168.97,-74.63) | - | - |
| 10 | 0 | - | - | - | - | (-177.11,-90.33) |
Table 3: Minimum energy conformations of neuraminic acid analogues with multiple Modifications in aqueous environment.
Contribution of hydrogen bonds in structural stability
Figs. 1-3 showed the formation of water mediated hydrogen bonds and direct hydrogen bonds in neuraminic acid derivatives. It is well known from the previous findings that water mediated hydrogen bonds formed within the molecule plays a major role in stabilization of the molecule[11]. The watermediated hydrogen bonds and direct hydrogen bonds are shown in Table 4. Comparison of water mediated hydrogen bonds with direct hydrogen bonds revealed that analogue 10 (5-N-acetyl-9-amino-9-deoxy neuraminic acid) has 4 water mediated hydrogen bonds and 4 direct hydrogen bonds. The nitrogen atom from the substituted NH2 group contributed to both water mediated and direct hydrogen bond. It is also observed that analogue 6 (2α-O-methyl-4-Ocapriloyl- 5-N-acetyl neuraminic acid) has three water mediated hydrogen bonds and three direct hydrogen bonds. It is inferred that the maximum number of direct and water mediated hydrogen bonds present in each neuraminic acid derivatives are responsible for the minimum energy and its stabilization of the molecule.

Figure 1: The 3D structure of neuraminic acid analogues in global
minimum energy conformation state.
(a) N-acetyl neuraminic acid. (b) methyl 5-N-acetyl neuraminate
(c) methyl 2α-O-methyl-5-N-acetyl neuraminate (d) benzyl 2α-Omethyl-
5-N-acetyl-8,9-O-isopropylidene.

Figure 2: The 3D structure of neuraminic acid analogues in global
minimum energy conformation state.
(a) benzyl 2α-O-methyl-4-O-capriloyl-5-N-acetyl-8,9-Oisopropylidene
neuraminate. (b) benzyl 2α-O-methyl-4-O-capriloyl-
5-N-acetyl-8,9-O-isopropylidene neuraminate (c) 2α-O-methyl-4-Ocapriloyl-
5-N-acetyl neuraminic acid (d) benzyl-2α-O-methyl-4-O-(8-
morpholin)-capriloyl-5-N-acetyl-8,9-O-isopropylidene-neuraminate.

Figure 3: The 3D structure of neuraminic acid analogues in global
minimum energy conformation state.
(a) benzyl-2α-O-methyl-4-O-(8-morpholin)-capriyloyl-5-N-acetylneuraminate.
(b) 2α-O-methyl-4-O-(8-morpholin)-capriloyl-5-Nacetyl
neuraminic acid (c) N-acetyl-9-amino-9-deoxy neuraminic acid.
| Sialic acid derivative | Interacting sialic acid derivative atom 1 | Mediating water | Distance (Å) | Interacting sialic acid derivative atom2 | Distance (Å) |
|---|---|---|---|---|---|
| N-acetyl Neuraminic Acid | O10 | WAT:180 | 2.77 | O4 | 2.97 |
| O2 | WAT:9 | 2.91 | O8 | 3.01 | |
| O1B | WAT:5 | 3.19 | O9 | 3.01 | |
| O9 | WAT:62 | 3.10 | O7 | 3.08 | |
| O8 | O7 | 3.01 | |||
| O2 | O1B | 2.85 | |||
| O4 | N5 | 2.93 | |||
| methyl 5-N-acetyl neuraminate | O10 | WAT:131 | 2.88 | O7 | 2.74 |
| O2 | WAT:165 | 3.17 | O7 | 3.10 | |
| O4 | N5 | 2.81 | |||
| O2 | O1B | 2.79 | |||
| methyl 2α-O-methyl-5-N-acetyl | O7 | WAT:176 | 2.60 | O10 | 3.07 |
| neuraminate | O1A | WAT:74 | 2.85 | O10 | 3.03 |
| O2 | O1B | 2.82 | |||
| O8 | O9 | 3.02 | |||
| O8 | O7 | 2.96 | |||
| O4 | N5 | 2.99 | |||
| benzyl 2α-O-methyl-5-N-acetyl-8,9-O- | O7 | WAT:19 | 2.67 | O9 | 3.13 |
| isopropylidene | O7 | WAT:2 | 3.14 | O2 | 2.81 |
| neuraminate | O2 | O1B | 2.94 | ||
| O4 | N5 | 3.01 | |||
| O8 | O9 | 3.21 | |||
| benzyl 2α-O-methyl-4-O-capriloyl- | O10 | WAT:181 | 2.90 | O11* | 3.17 |
| 5-N-acetyl-8,9-O-isopropylidene | O9 | WAT:121 | 2.86 | O2 | 2.88 |
| neuraminate | N5 | O4 | 2.92 | ||
| O8 | O7 | 2.81 | |||
| benzyl 2α-O-methyl-4-O-capryloil-5- | O2 | WAT:193 | 3.03 | O9 | 2.71 |
| N-acetyl neuraminate | O10 | WAT:126 | 2.48 | O11 | 3.19 |
| O7 | O8 | 2.91 | |||
| O8 | O9 | 2.90 | |||
| O2 | O1B | 2.80 | |||
| N5 | O4 | 2.87 | |||
| 2α-O-methyl-4-O-capriloyl-5-N-acetyl | O7 | WAT:75 | 2.59 | O10 | 2.81 |
| neuraminic acid | O10 | WAT:169 | 2.87 | O11* | 2.80 |
| O1A | WAT:71 | 3.20 | O9 | 3.07 | |
| N5 | O4 | 2.82 | |||
| O2 | O1B | 3.06 | |||
| O7 | O8 | 2.95 | |||
| benzyl-2α-O-methyl-4-O-(8- | O8 | WAT:166 | 2.65 | O2 | 3.08 |
| morpholin)-capriloyl-5-N-acetyl-8,9- | N18* | O20* | 2.87 | ||
| O-isopropylidene-neuraminate | N5 | O4 | 2.86 | ||
| O2 | O1B | 2.84 | |||
| O7 | O8 | 2.98 | |||
| benzyl-2α-O-methyl-4-O-(8- | O1B | WAT:199 | 3.07 | O4 | 2.93 |
| morpholin)-capriyloyl-5-N-acetyl- | O11* | WAT:198 | 3.03 | O10 | 2.42 |
| neuraminate | O8 | O7 | 2.88 | ||
| O9 | O8 | 3.13 | |||
| O2 | O1B | 2.90 | |||
| N5 | O4 | 2.78 | |||
| N18* | O20* | 2.88 | |||
| 2α-O-methyl-4-O-(8-morpholin)- | O9 | WAT:82 | 3.04 | O7 | 2.48 |
| capriloyl-5-N-acetyl neuraminic acid | O9 | WAT:81 | 2.89 | O1A | 2.83 |
| N18* | O20* | 2.88 | |||
| O8 | O7 | 2.96 | |||
| O2 | O1B | 2.87 | |||
| O4 | N5 | 2.85 | |||
| 5-N-acetyl-9-amino-9-deoxy | O1B | WAT:44 | 2.75 | N9* | 3.10 |
| neuraminic acid | O10 | WAT:177 | 3.14 | O7 | 2.90 |
| O4 | WAT:193 | 3.04 | O2 | 2.68 | |
| O5 | WAT:146 | 2.71 | O10 | 2.89 | |
| O8 | N9* | 2.52 | |||
| O7 | O8 | 3.21 | |||
| N5 | O4 | 3.05 | |||
| O2 | O1B | 2.81 |
*is the atom from the substituent group.
Table 4: Hydrogen bonds in each neuraminic acid analogue with multiple modifications.
Molecular dynamics of sialic acid analogues
To study the conformational dynamics of the neuraminic acid derivatives, a 2ns molecular dynamics simulation was carried out. An in-depth analysis on the conformational features of all the 10 neuraminic acid analogues was done by collecting the frames for every 1ps.
C-1 substituted side chain conformation
For analogue 1 (methyl 5-N-acetyl neuraminate), the dihedral angles γ1 and γ2 exhibit a bifurcation throughout the molecular dynamics simulation. The torsional angle γ1 of analogue 2 (methyl 2α-O-methyl- 5-N-acetyl neuraminate), analogue 3 (benzyl 2α-Omethyl- 5-N-acetyl-8,9-O-isopropylidene neuraminate), analogue 4 (benzyl 2α-O-methyl-4-O-capriloyl-5-Nacetyl- 8,9-O-isopropylidene neuraminate), analogue 5 (benzyl 2α-O-methyl-4-O-capryloil-5-N-acetyl neuraminate) and analogue 8 (benzyl-2α-O-methyl-4- O-(8-morpholin)-capriyloyl-5-N-acetyl-neuraminate) shows good distribution throughout the MD simulation, however γ2 is rigid in +180° and -180° regions. The shift of γ1 from -180° region to +70° region results in the energy decrease of 5 kcal/mol. Fig. 4a shows the molecular dynamic trajectory showing transitions of the substituent holding side chain torsion angles γ1 and γ2 along with (γ1, γ2) distribution plot of analogue 5 (benzyl 2α-O-methyl-4-O-capryloil-5-N-acetyl neuraminate). In the case of analogue 7 (benzyl-2α-Omethyl- 4-O-(8-morpholin)-capriloyl-5-N-acetyl-8,9-Oisopropylidene- neuraminate), γ1 bifurcates in negative regions such as -180°, -120° and -60°, whereas γ2 prefers +180° and -180° region.

Figure 4: Molecular dynamics trajectory and the distribution plots of
sialic acid analogues
(a) γ1 Vs γ2 plots for benzyl 2α-O-methyl-4-O-capryloil-5-N-acetyl
neuraminate, (b) θ1 Vs θ2 distribution plots for 2α-O-methyl-4-Ocapriloyl-
5-N-acetyl neuraminic acid (c) β1 Vs β2 plots for 2α-O-methyl-
4-O-capriloyl-5-N-acetyl neuraminic acid.
C-2 substituted side chain conformation
For analogue 2 (methyl 2α-O-methyl-5-N-acetyl neuraminate), 3 (benzyl 2α-O-methyl-5-N-acetyl- 8,9-O-isopropylidene neuraminate), 4 (benzyl 2α-Omethyl- 4-O-capriloyl-5-N-acetyl-8,9-O-isopropylidene neuraminate), 5 (benzyl 2α-O-methyl-4-O-capryloil-5- N-acetyl neuraminate), 6 (2α-O-methyl-4-O-capriloyl- 5-N-acetyl neuraminic acid) and 7 (benzyl-2α-Omethyl- 4-O-(8-morpholin)-capriloyl-5-N-acetyl-8,9- O-isopropylidene-neuraminate), θ1 is rigid in -60° region and θ2 prefers +180° and -180° regions. Fig. 4b represents the dynamic behavior of torsions θ1and θ2 along with (θ1,θ2) distribution plot of 2α-O-methyl-4- O-capriloyl-5-N-acetyl neuraminic acid (analogue 6) in aqueous environment.
However the dihedral angle θ2 of analogue 3 (benzyl 2α-O-methyl-5-N-acetyl-8,9-O-isopropylidene neuraminate), analogue 5 (benzyl 2α-O-methyl-4-Ocapryloil- 5-N-acetyl neuraminate) and analogue 6 (2α-O-methyl-4-O-capriloyl-5-N-acetyl neuraminic acid) exhibits an additional bifurcation in -60° region. In the case of analogue 8 (benzyl-2α-O-methyl-4-O- (8-morpholin)-capriyloyl-5-N-acetyl-neuraminate), θ1 prefers -70° region and θ2 prefers +180°, -180° and -70° regions.
C-4 substituted side chain conformation
β1 of analogue 4 (benzyl 2α-O-methyl-4-O-capriloyl-5- N-acetyl-8,9-O-isopropylidene neuraminate), 5 (benzyl 2α-O-methyl-4-O-capryloil-5-N-acetyl neuraminate), 7 (benzyl-2α-O-methyl-4-O-(8-morpholin)-capriloyl- 5-N-acetyl-8,9-O-isopropylidene-neuraminate), and 8 (benzyl-2α-O-methyl-4-O-(8-morpholin)-capriyloyl- 5-N-acetyl-neuraminate) prefers +180° and -180° regions. β2 of analogue 5 (benzyl 2α-O-methyl-4- O-capryloil-5-N-acetyl neuraminate), 7 (benzyl-2α- O-methyl-4-O-(8-morpholin)-capriloyl-5-N-acetyl- 8,9-O-isopropylidene-neuraminate) and 8 (benzyl-2 α-O-methyl-4-O-(8-morpholin)-capriyloyl-5-N-acetylneuraminate) prefers -180° and -70° regions. β2 of analogue 4 (benzyl 2α-O-methyl-4-O-capriloyl-5-Nacetyl- 8,9-O-isopropylidene neuraminate) prefers -180°, -120°, +180° and -60° regions.
For analogue 7 (benzyl-2α-O-methyl-4-O-(8- morpholin)-capriloyl-5-N-acetyl-8,9-O-isopropylideneneuraminate), the shift of β2 from -70° to -180° region results in the energy decrease of up to 6 kcal/ mol. In the case of analogue 6 (2α-O-methyl-4-Ocapriloyl- 5-N-acetyl neuraminic acid), β2 shows good distribution in -180°, +180°, -70° and +60° regions. β1 prefers +180° and -180° regions. Fig. 4c represents the dynamic behavior of torsions β1 and β2 along with (β1, β2) distribution plot of 2α -O-methyl-4-O-capriloyl- 5-N-acetyl neuraminic acid (analogue 6) in aqueous environment.
C-8 substituted side chain conformation
For analogue 3 (benzyl 2α-O-methyl-5-N-acetyl- 8,9-O-isopropylidene neuraminate), analogue 4 (benzyl 2α-O-methyl-4-O-capriloyl-5-N-acetyl-8,9-Oisopropylidene neuraminate) and analogue 7 (benzyl- 2α-O-methyl-4-O-(8-morpholin)-capriloyl-5-N-acetyl- 8,9-O-isopropylidene-neuraminate), χ1 prefers +180° and -180° regions and χ3 is rigid in +70° region. Fig. 5a describes the molecular dynamic trajectory showing transitions of the different substituent holding side chain torsion angles χ1 and χ3 along with (χ1, χ3) distribution plot of analogue 3 (benzyl 2α-O-methyl-5- N-acetyl-8,9-O-isopropylidene neuraminate).

Figure 5: Molecular dynamics trajectory and the distribution plots (a) χ1 Vs χ3 plots for benzyl 2α-O-methyl-5-N-acetyl-8,9-Oisopropylidene neuraminate, (b) χ1 Vs χ2 plots for benzyl 2α-Omethyl- 4-O-capriloyl-5-N-acetyl-8,9-O-isopropylidene neuraminate and (c) χ1 Vs χ2 plots for 5-N-Acetyl-9-amino-9-deoxy neuraminic acid
C-9 substituted side chain conformation
χ1 analogue 3 (benzyl 2α-O-methyl-5-N-acetyl-8,9- O-isopropylidene neuraminate), 4 (benzyl 2α-Omethyl- 4-O-capriloyl-5-N-acetyl-8,9-O-isopropylidene neuraminate) and 7 (benzyl-2α-O-methyl-4-O-(8- morpholin)-capriloyl-5-N-acetyl-8,9-O-isopropylidene neuraminate) prefers +180° and -180° regions and χ2 prefers -70° region. In the case of analogue 10 (5-N-acetyl-9-amino-9-deoxy neuraminic acid), χ1 prefers -180° +60° and +180° regions and χ2 shows bifurcation in -70°, +70°, +180° and -180° regions. The shift of χ1 from -180° region to +60° region resulted in the energy decrease of up to 10kcal/mol. Fig. 5 (b and c) represents the dynamic behavior of torsions χ1 and χ2 along with (χ1, χ2) distribution plot of neuraminic acid analogues benzyl 2α-O-methyl-4-O-capriloyl-5- N-acetyl-8,9-O-isopropylidene neuraminate (analogue 4) and 5-N-acetyl-9-amino-9-deoxy neuraminic acid (analogue 10) in aqueous environment.
Modeling of sialic acid analogues and Influenza hemagglutinin complexes
High Throughput Virtual Screening was done for ten neuraminic acid analogues to find out the structures (ligands) most likely to bind to the Influenza hemagglutinin. The top fi ve ligands with best docking score and minimum energy are subjected to induced fi t docking. The glide score along with the glide energy is displayed in Table 5. The inter-molecular interactions between the top five ligands and the Influenza hemagglutinin are noted and displayed in Table 6. Figs. 6 and 7 shows the neuraminic acid analogues 3 (benzyl 2α-O-methyl-5-N-acetyl-8,9-O-isopropylidene neuraminate) and 10 (5-N-acetyl-9-amino-9-deoxy neuraminic acid) at the active site of Influenza HA, respectively. The bound state conformations of the substituent holding side chains of sialic acid analogues are displayed in Table 7.
| Analogue no | Neuraminic acid derivative | Glide energy (kcal/mol) | Glide score |
|---|---|---|---|
| 5 | Benzyl 2a-O-methyl-4-O-capryloil-5-N-acetyl Neuraminate | -54.64 | -8.64 |
| 10 | 5-N-Acetyl-9-amino-9-deoxy Neuraminic Acid | -36.70 | -7.73 |
| 2 | 5-N-Acetyl-9-amino-9-deoxy Neuraminic Acid | -42.87 | -7.71 |
| 4 | Benzyl 2α-O-methyl-4-O-capriloyl-5-N-acetyl-8,9-O-isopropylidene Neuraminate | -45.91 | -6.71 |
| 3 | Benzyl 2a-O-methyl-5-N-acetyl-8,9-O-isopropylidene Neuraminate | -42.16 | -5.34 |
Table 5: Glide docking score and glide energy of influenza hemagglutinin - neuraminic acid complexes.

Figure 6: Sialic acid analogue (2α-O-methyl-5-N-acetyl-8,9-Oisopropylidene neuraminate) at the active site of Influenza Hemagglutinin.

Figure 7: Sialic acid analogue (5-N-acetyl-9-amino-9-deoxy neuraminic acid) at the active site of Infl uenza Hemagglutinin.
| Analogue no | Neuraminic acid derivative | Ligand atom | Protein | Distance | |
|---|---|---|---|---|---|
| Residue | Atom | (Å) | |||
| 5 | Benzyl 2a-O-methyl-4-O-capryloil-5-N-acetyl Neuraminate | O8 | GLU:225 | OE1 | 2.677 |
| O9 | GLU:225 | OE1 | 2.698 | ||
| O9 | GLY:227 | N | 2.338 | ||
| 10 | 5-N-Acetyl-9-amino-9-deoxy Neuraminic Acid | N | GLY:134 | O | 3.061 |
| O1 | GLY:134 | O | 2.748 | ||
| N9 | ASN:185 | OD1 | 3.261 | ||
| N | GLU:189 | OE1 | 2.913 | ||
| O10 | GLN:225 | OE1 | 2.901 | ||
| 2 | Methyl 2α-O-methyl-5-N-acetyl Neuraminate | O9 | GLY:134 | O | 2.798 |
| O2 | GLU:189 | OE1 | 2.677 | ||
| O1 | TYR:95 | OH | 2.675 | ||
| 3 | Benzyl 2a-O-methyl-5-N-acetyl-8,9-O-isopropylidene Neuraminate | O7 | GLU:189 | OE1 | 2.830 |
| 4 | Benzyl 2α-O-methyl-4-O-capriloyl-5-N-acetyl-8,9-O-isopropylidene | O9 | GLU:225 | OE1 | 2.743 |
| Neuraminate | O7 | GLU:225 | OE1 | 2.991 | |
| O4 | GLY:134 | O | 2.936 | ||
Table 6: Inter-molecular interactions between the neuraminic acid analogues and influenza hemagglutinin.
| Neuraminic acid Analogue No | C1 substitution (γ1, γ2) (deg.) | C2 substitution (θ1,θ2) (deg.) | C4 substitution (β1, β2) (deg.) | C8 substitution (χ1, χ3) (deg.) | C9 substitution (χ1, χ2) (deg.) |
|---|---|---|---|---|---|
| - | - | - | - | - | |
| 2 | (47.8,-179.9) | (-66.4,-178.7) | - | - | - |
| 3 | (92.0,171.9) | (-74.7,166.5) | - | (102.9,-63.0) | (102.9,-176.5) |
| 4 | (48.1,179.7) | (-63.7,-54.1) | (-178.0,-74.1) | (-171.4,50.3) | (-171.4,-75.0) |
| 5 | (13.8,158.0) | (-66.1,-138.7) | (-172.8,-63.6) | - | - |
| 10 | - | - | - | - | (-173.5,96.1) |
Table 7: Bound state conformations of the substituent holding side chains of sialic acid Analogues.
Discussion
The current study reveals the probable conformational models for neuraminic acid derivatives with multiple substitutions at positions C-1/C-2/C-4/C-8/C-9 in aqueous environment. Water mediated hydrogen bonding interaction plays a dominant role in stabilizing the conformational structures of these neuraminic acid derivatives. The accessible conformations for neuraminic acid analogues with multiple substituents holding side chain linkages observed by the present MD study correlate well with those reported for similar linkages in various Neu5Ac-α2→8-Neu5Ac moiety present in all the di- and tri-sialogangliosides by earlier studies[11,12]. Present MD results show a dynamic behaviour for χ1 of analogue 10 (5-N-acetyl- 9-amino-9-deoxy neuraminic acid) at the cost of 10 kcal/mol (Fig. 5c), which is a vivid indication that this molecule prefers -60° region. Analogue 10 is observed to have the lowest energy of -2395.1 kcal/mol when compared to other neuraminic acid analogues with multiple substituents and it shows good results in hydrogen bonding interactions with four water mediated hydrogen bonds and four direct hydrogen bonds. The nitrogen from the substituted NH2 group contributes to both water mediated and direct hydrogen bond. Docking studies show the mode of binding of sialic acid derivatives into the binding pocket of Influenza hemagglutinin which in turn explains their order of specifi city. It is very interesting to know that the neuraminic acid analogues, benzyl 2α -O-methyl-4-O-capryloil-5-N-acetyl neuraminate (analogue 5), 5-N-acetyl-9-amino-9-deoxy neuraminic acid (analogue 10), 5-N-acetyl-9-amino-9-deoxy neuraminic acid (analogue 2), benzyl 2α-O-methyl-4-Ocapriloyl- 5-N-acetyl-8,9-O-isopropylidene neuraminate (analogue 4) and benzyl 2α-O-methyl-5-N-acetyl- 8,9-O-isopropylidene neuraminate (analogue 3) have greater docking score than the co-crystal ligand (PDB reference ligand) such as -8.64, -7.73, -7.71, -6.71 and -5.34, respectively and minimum energy such as -54.64 kcal/mol, -36.70 kcal/mol, -42.87 kcal/mol, -45.91 kcal/ mol and -42.16 kcal/mol, respectively.
The present study provides accessible conformational models for synthetic neuraminic acid analogues with multiple substituents at positions C-1, C-2, C-4, C-8 or C-1 in aqueous environment. Direct and water mediated hydrogen bonding schemes greatly involve in stabilizing the three dimensional conformational structures of these neuraminic acid analogues. This study also shows the dynamics trajectory and distribution plot for the substituent holding side chain linkages of the neuraminic acid analogues. The high affinity inhibitors modeled in this study saturate the hemagglutinin (HA) receptor[5] and can be used as potential antiinfl uenza drugs.
Acknowledgements
We thank Prof. Dr. K. Veluraja for helpful discussions.
References
- Paulson JC. Interactions of animal viruses with cell surface receptors. Receptor 1985;2:131-219.
- Wiley DC, Skehel JJ. The structure and function of hemagglutinin membrane glycoprotein of influenza virus. Ann Rev Biochem 1987;56:365-94.
- Weis W, Brown JH, Cusack S, Paulson JC, Skehel JJ, Wiley DC. Structure of the influenza virus hemagglutinincomplexed with its receptor, sialic acid. Nature 1988;33:426-31.
- Kelm S, Paulson JC, Rose U, Brossmer R, Schmid W, Bandgar BP, etal. Use of sialic acid analogues to define functional groups involvedin binding to the influenza virus hemagglutinin. Eur J Biochem 1992;205:147-53.
- Sauter NK, Hanson JE, Glick GD, Brown JH, Crowther RL, Park SJ, etal. Binding of influenza virus hemagglutinin to analogs of its cell-surfacereceptor, sialic acid: Analysis by proton nuclear magnetic resonance spectroscopy and X-ray crystallography. Biochemistry 1992;31:9609-21.
- Bianco BM, Ciabatti R, Melchioni C, Pasquali V. Neuraminic acid derivatives as anti-influenza drugs. Mol Online 1988;2:129-36.
- Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, etal. The Amber biomolecular simulation programs. J ComputChem2005;26:1668-88.
- Rollinger JM, Stuppner H, Langer T. Virtual screening for the discovery of bioactive natural products. Prog Drug Res 2008;65:213-49.
- Walters WP, Stahl MT, Murcko MA. Virtual screening: An overview. Drug Discov Today 1998;3:160-78.
- Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem 2006;49:534-53.
- Sharmila DJ, Veluraja K. Conformations of higher gangliosides and their binding with cholera toxin-investigation by molecular modeling, molecular mechanics and molecular dynamics. J BiomolStructDyn 2006;23:641-56.
- Vasudevan SV, Balaj PV. Comparitive analysis of ganglioside conformations by MD simulations: Implications for specific recognition by proteins. J MolStruct 2001;583:215-32.





