- *Corresponding Author:
- Zihong Yan
Department of Chemistry, College of Chemistry and Environmental Science, Kashgar University, Kashgar, Xinjiang 844006, China
E-mail: yanzi0928@126.com
| This article was originally published in a special issue, “Recent Developments in Biomedical Research and Pharmaceutical Sciences” |
| Indian J Pharm Sci 2022:84(4) Spl Issue “184-195” |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
In this study, the interaction between human immunodeficiency virus reverse transcriptase and methyldiarylpyrimidines containing a hydroxyimino, hydrazine, hydroxyl, cyclopropylamino, cyano or chloro etc. substituent was studied by molecular docking simulation, molecular docking was accomplished with AutoDock4.2 and absorption, distribution, metabolism, excretion and toxicity-structure-activity relationship and Swiss absorption, distribution, metabolism and excretion servers were used to predict the pharmacokinetics and absorption, distribution, metabolism, excretion and toxicity properties of all compounds. As a result, molecular docking analysis revealed that there are extensive interactions between the diarylpyrimidine derivatives and Lys101, Tyr188, Val179, Lys103, Glu138, Leu234, Phe227 and Trp229 residues in the active site of human immunodeficiency virus-1 reverse transcriptase. The formation of hydrogen bonds between diarylpyrimidines and the residues Lys101 and Glu138 play important roles in inhibiting the activity of human immunodeficiency virus. All compounds respect the conditions mentioned in Lipinski’s rule and have acceptable absorption, distribution, metabolism, excretion and toxicity properties. The preliminary structure-activity relationship was also investigated. A preliminary structureactivity relationship analysis of these target molecules highlighted a preference for the smaller or more flexible group’s substitution in the linker between the left ring and the pyrimidine skeleton for enhancing the anti-human immunodeficiency virus-1 activity. The acrylonitrile C4-substituent substantially improved the potency against wild-type human immunodeficiency virus-1 and several mutant strains, and the 2nd position is the most preferred position to improve antiviral activity. This information might be helpful for further diarylpyrimidine optimizations.
Keywords
Absorption, distribution, metabolism, excretion and toxicity, diarylpyrimidines, molecular docking, pharmacokinetics, structure-activity relationship
Human Immunodeficiency Virus type 1 (HIV-1) is the primary cause of Acquired Immunodeficiency Syndrome (AIDS), which is still a serious public health issue on a global scale[1]. HIV-1 Reverse Transcriptase (RT) converts the single-stranded Ribonucleic Acid (RNA) genome into a doublestranded Deoxyribonucleic Acid (DNA) copy, which is necessary for HIV-1 replication and has been identified as a promising therapeutic target for the development of anti-HIV drugs[2]. Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs) can be disrupted by NNRTIs, which can attach to a hydrophobic allosteric binding pocket (NNRTI Binding Pocket, NNIBP) that is situated at position 10 from the RT polymerase active site[3]. Components of Highly Active Antiretroviral Treatment (HAART) have a special antiviral potency[4].
Despite having a number of benefits, only four NNRTIs-Nevirapine (NVP), Delavirdine (DLV), Efavirenz (EFV) and Etravirine (ETV), have received clinical use approval from the Food and Drug Administration (FDA)[5]. First-generation NNRTIs include NVP, DLV and EFV, but their therapeutic efficacy has been dramatically understated due to the rapid emergence of HIV-1 drug resistant variants such as Tyr181, Lys103 and Tyr188 single mutants, as well as Lys103/Tyr181 double mutant (RES056 Lys103+Tyr181)[6]. In order to combat the AIDS epidemic, the United States (US) FDA approved the next-generation HIV-1 NNRTIs (ETV, TMC125, fig. 1) and Rilpivirine (RPV, TMC278, fig. 1) in 2008 and 2011, respectively[7].

Fig. 1: Chemical structures of DAPY-type NNRTI drugs, (A) ETV and (B) RPV
The diarylpyrimidine compounds are important in the non-nucleoside reverse transcriptase inhibitors, because it has a strong inhibitory activity and small toxic side effects on single mutation and double mutation. Diarylpyrimidines have been discovered via the bioisosterism principle or molecular hybridization concept. TMC125 and TMC278 are Diarylpyrimidine (DAPY) NNRTIs with high activity against Wild-Type (WT) and mutant virus strains including Lys103[8]. However, the resistant variants Glu138 and Lys103/ Tyr181, can still significantly decrease the sensitivity of HIV-1 to ETV and RPV[9]. Therefore, there is a need of developing novel NNRTIs with enhanced therapeutic value and different resistance mutation profiles to successfully employ this class of drugs in HAART[10]. A series of new NNRTIs has been found and synthesized in the ongoing study of NNRTIs in the last decade. DAPY is one of them and a design feature for these compounds is the innate flexibility between aromatic rings, allowing the compound to adopt multiple conformations and is one explanation for the potent activity against many resistant virus strains (fig. 2). So it has a promising prospect of development and application[11].

Fig. 2: The general structure of DAPYs
In this study, we have taken 10 DAPYs that displayed the most potent anti-HIV-1 activity, with excellent selectivity for infected over uninfected cells. DAPYs, 1[12], 2[13], 3[14] 4[15], 5[16], 6-7[17], 8[18], 9[19] and 10[20] studied in silico for their potential anti-HIV activity along with their physicochemical and Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) properties. The relative Structure-Activity Relationship (SAR) discussions were also investigated together with the comparative docking analysis. The principles of medicinal chemistry and bioinformatics tools help to locate effective therapeutics to minimize resources and time in current drug development modules.
Materials and Methods
The SwissADME[21] and admetSAR[22] servers were used to analyze the pharmacokinetic properties of DAPYs. The Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) was used to obtain protein crystal structures based on resolution and R factors, specifically 2ZD1 and 3BGR. AutoDock tools 1.5.6 were used to prepare the proteins[23] to remove unwanted co-crystallized molecules, to add charges (Gaussian, Gasteiger) and to incorporate polar hydrogens. Their energy minimized ligand data was developed on ChemDraw. Blind docking was achieved using AutoDock tools 1.5.6 to assist. Post-docking analysis of protein-ligand interactions was conducted on BIOVIA Discovery Studio Visualizer 2021.
Drug-likeness properties:
Pharmacokinetics is the study of the time course of a drug within the body and incorporates the processes of ADME. Pharmacokinetic parameters are derived from the measurement of drug concentrations in blood or plasma. Most drugs are given orally for reasons of convenience and compliance. Typically, a drug dissolves in the gastro-intestinal tract is absorbed through the gut wall and then passes the liver to get into the blood circulation. The percentage of the dose reaching the circulation is called the bioavailability. From there, the drug will be distributed to various tissues and organs in the body. The extent of distribution will depend on the structural and physicochemical properties of the compound. Some drugs can enter the brain and central nervous system by crossing the Blood-Brain Barrier (BBB). Finally, the drug will bind to its molecular target. For example, a receptor or ion channel exerts its desired action.
The compounds chosen from structure-based virtual screening were evaluated for drug-likeness and Lipinski’s “rule of five”[24] parameters, which included molecular weight, number of Hydrogen (H)-bond donors, number of H-bond acceptors and partition coefficient[24]. A drug or compound that is taken orally should have a molecular mass of 500 Daltons (Da), calculated octanol/water partition coefficient (CLogP)<5, number of hydrogen-bond donors<5 and number of hydrogen-bond acceptors<10. These properties are then typically used to construct predictive ADME models and form the basis for what has been called property-based design. To a certain extent, similar molecules can be expected to have similar ADME properties. While Veber et al. suggested that a molecule or medicine should have a Topological Polar Surface Area (TPSA) value of 1402 and a number of rotatable bonds of atleast 10. All of these guidelines must be followed in order for a medicine or chemical to be deemed to have good oral bioavailability. Using the online SwissADME calculator, all of these physicochemical characteristics and drug-likeness factors were determined[25].
ADMET properties:
Potential therapeutic candidate’s pharmacokinetic properties, or ADMET assessments, describe how these substances are disposed of within an organism. A drug success is dependent on both of its effectiveness and its ability to provide appropriate ADMET descriptors[26]. As a result, online admetSAR was used to predict the carcinogenicity, mutagenicity, irritant, reproductive impacts and ADMET characteristics of 10 compounds that were anticipated to be active. AdmetSAR employs a proprietary substructure pattern recognition technique and a vector machine classification algorithm. In this server, the database can be searched by common name, Chemical Abstracts Service Registry Number (CASRN), International Union of Pure and Applied Chemistry (IUPAC) name, Simplified Molecular Input Line Entry System (SMILES) and structural similarity[22]. Thanks to web-based query tools with a molecular built-in interface. Different toxicity metrics, interactions between metabolizing enzymes and absorption factors were assessed, and substances were checked for its drug-like characteristics.
Molecular docking simulation:
Using the software AutoDock 4.2, this set of compounds SAR data and binding manner were better understood[27]. Discovery Studio Visualizer 2021 was applied for molecular graph display and result analysis. The crystal structure WT RT (PDB code: 2ZD1) and the Lys103/Tyr181 double-mutant RT (PDB: 3BGR) with the same analogue was retrieved from the Book Haven PDB.
First, the DAPYs micromolecular structure was drawn by the ChemDraw 19.1 software and geometric optimization was carried out. Second, the crystallographic structure of HIV-1 protease is imported into “work space” of Discovery Studio Visualizer 2021 program to obtain the binding site. The center of the active site has been determined and it corresponds to the coordinates: x, y, z on the basis of the co-crystallized bound peptidomimetic ligand (Table 1). Third, the receptor protein was analyzed by AutoDock4.2 and receptors were processed by AutoDock tools to eliminate water and other ligands, additional charge and additional hydrogen. Automatic detection rigid atoms were used to process ligand micromolecules. All torsional bonds and torsional trees are flexible. In the AutoGrid program, the grid interval in the gridding map was set at the default value of 0.375 Å and grid size of 40 Å×40 Å×40 Å. During docking, images were searched by the Lamarckian genetic algorithm to study binding sites between receptors and ligands. Finally, the hydrophobic interaction force was analyzed by using the Discovery Studio Visualizer 2021 software.
| Complex PDB ID | Ligand ID | Chemical structure | RMSD (Å) | Binding sites | Resolution | ||
|---|---|---|---|---|---|---|---|
| x | y | z | |||||
| 2ZD1 | RPV, TMC278 |
|
0.375 Å | 50.4819 | -27.515 | 37.0398 | 1.80 Å |
| 3BGR | 50.1345 | -27.888 | 36.7754 | 2.10 Å | |||
Note: RMSD: Root-Mean-Square Deviation; PDB: Protein Data Bank and RPV: Rilpivirine
Table 1: Redocking of References Ligand Against HIV-1 Complexes
Results and Discussion
Pharmacokinetic investigation of DAPYs was explained here. Pharmacokinetic properties are an elemental segment of drug development to identify the biological properties of drug candidates. Lipinski’s rule of five[24] and Veber’s rules[25] were used to check the drug-likeness. DAPYs given in Table 2 were analyzed for their physicochemical properties and the results are epitomized in Table 3. The ADMET characteristics of the DAPYs were studied to understand their pharmacokinetic profile and the results are given in Table 4.
| N | Chemical structure | EC50 µmol/l | RES056 EC50 (µM]) |
N | Chemical structure | EC50 µmol/l | RES056 EC50 (µM) |
||
|---|---|---|---|---|---|---|---|---|---|
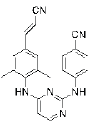
| CH (NOH)-DAPY | 1 |
|
0.005 | >13.23 | 7 | 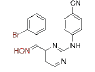 |
0.065 | 8.8±0.5 | |
| CH (NNH2)-DAPY | 2 | 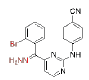 |
0.0017 | ≥9.2 | C (Me) (OH)-DAPYs |
8 | 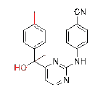 |
0.067 | >39.9 |
| 3 | 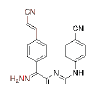 |
0.0018 | 5.33 | CHX-DAPYs | 9 | 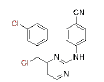 |
0.005 | >24.3 | |
| CH (Cpa)-DAPYs | 4 | 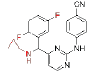 |
0.099 | >228.941 | CH (CN)-DAPYs | 10 | 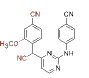 |
0.059 | >25.85 |
| CH (OH)-DAPY | 5 | 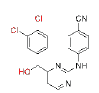 |
0.008 | >1.8 | ETV | 0.0055 | 0.04 | ||
| 6 | 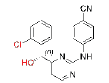 |
0.005 | >36 | PRV | 0.0016 | 0.009 |
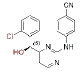
Table 2: The Structures and Bioactivity Values of Activity of DAPYs
Out of 10 DAPYs listed in Table 2, all DAPYs are showing no violation of the Lipinski’s and Veber’s rules and hence display Drug-Like Molecular (DLM) nature. The Log P values of DAPYs are within the range of 2.99 to 3.99. Molecular weight, number of H-acceptors and H-bond donors of all DAPYs are within the accepted values of less than 500, 10 and 5 respectively (Table 3) and all the DAPYs are following the criteria of Veber’s rules with Total Polar Surface Area (TPSA) values and the count of rotatable bonds within range for oral availability.
| Name | Lipinski’s rule of 5 | Veber’s rule | |||||
|---|---|---|---|---|---|---|---|
| LogP | Molecular Weight | Hydrogen donor | Hydrogen acceptor | Number of violations | TPSA (Å2) | Number of rotatable bonds | |
| 1 | 3.41 | 394.22 | 2 | 5 | 0 | 94.19 | 4 |
| 2 | 3.01 | 393.24 | 2 | 4 | 0 | 99.98 | 4 |
| 3 | 2.65 | 365.39 | 2 | 5 | 0 | 123.77 | 5 |
| 4 | 3.78 | 377.39 | 2 | 6 | 0 | 73.63 | 6 |
| 5 | 3.43 | 371.22 | 2 | 4 | 0 | 81.83 | 4 |
| 6 | 3.00 | 336.78 | 2 | 4 | 0 | 81.83 | 4 |
| 7 | 2.99 | 336.78 | 2 | 4 | 0 | 81.83 | 4 |
| 8 | 3.1 | 230.38 | 2 | 4 | 0 | 81.83 | 4 |
| 9 | 3.99 | 355.22 | 1 | 3 | 0 | 61.6 | 4 |
| 10 | 2.61 | 366.38 | 1 | 6 | 0 | 118.41 | 5 |
Table 3: Physicochemical Properties of DAPYs Under Study
| Name | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ETV | RPV |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BBB | 0.7066 | 0.732 | 0.8121 | 0.7445 | 0.8127 | 0.8127 | 0.813 | 0.7636 | 0.9285 | 0.9197 | 0.9173 | 0.8571 |
| HIA | 0.8323 | 0.9565 | 0.9843 | 0.9847 | 0.9825 | 0.9825 | 0.983 | 0.9821 | 0.9881 | 0.9939 | 0.9902 | 0.9929 |
| Caco-2 | 0.5358 | 0.7063 | 0.6647 | 0.602 | 0.6518 | 0.6518 | 0.652 | 0.6036 | 0.7262 | 0.7226 | 0.5 | 0.6609 |
| P-CPS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS |
| P-GPI | NI | NI | NI | NI | NI | NI | NI | NI | NI | NI | NI | NI |
| Renal organic cation transporter | NI | NI | NI | NI | NI | NI | NI | NI | NI | NI | NI | NI |
| CYP450 2C9 substrate | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS |
| CYP450 2D6 substrate | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS |
| CYP450 3A4 substrate | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS |
| CYP450 1A2 inhibitor | I | I | I | NI | I | I | I | I | I | I | I | I |
| CYP450 2C9 inhibitor | NI | NI | NI | NI | NI | NI | NI | NI | NI | NI | NI | NI |
| AMES toxicity | NT | NT | T | NT | NT | NT | NT | NT | NT | T | T | NT |
| Carcinogens | NC | NC | NC | NC | NC | NC | NC | NC | NC | NC | NC | NC |
Note: Blood-Brain Barrier (BBB): Don’t cross BBB (–); cross BBB (+); Human Intestinal Absorption (HIA): Not absorbed (–); absorbed (+), Caco-2 permeability: Not permeable (–); permeable (+); CYP450: S=Substrate for enzyme; NS=Not a substrate for enzyme; I=Enzyme inhibitor; NI=Not enzyme inhibitor; NT: Nontoxic; T: Toxic and NC: Noncarcinogenic
Table 4: Admet Properties of DAPYs
Molecular docking was carried out using the AutoDock program in order to explore the binding manner of DAPYs in the NNIBP of WT HIV-1 RT. To determine the kinds of interactions and the binding affinities of the investigated compounds in the investigated enzyme, molecular docking was used. 10 different DAPYs compounds have been evaluated for their affinity against the HIV RT protease. The results are presented in Table 5. The lowest binding free energy between HIV RT and DAPYs was calculated on the basis of the molecular docking simulation as -11.96 (2ZD1) and -11.37 (3BGR) kcal/mol.
| PDB | Ligand | Binding energy (kcal/mol) |
|---|---|---|
| 2ZD1 | 1 | -10.8 |
| 2 | -10.78 | |
| 3 | -11.96 | |
| 4 | -9.92 | |
| 5 | -10.33 | |
| 6 | -9.88 | |
| 7 | -9.97 | |
| 8 | -10.04 | |
| 9 | -10.94 | |
| 10 | -11.22 | |
| 3BGR | 1 | -10.45 |
| 2 | -10.32 | |
| 3 | -11.37 | |
| 4 | -9.89 | |
| 5 | -10.16 | |
| 6 | -9.69 | |
| 7 | -9.63 | |
| 8 | -9.73 | |
| 9 | -10.66 | |
| 10 | -10.95 |
Table 5: Receptor Ligand Interactions Between 3MEC 1S6Q 2ZD1 and 3BGR and DAPYs Compounds
We also assessed the in silico ADMET properties of two well-known drugs to validate the in silico protocol (ETV and RPV). The findings showed that every substance tested, including ETV and RPV, could pass through the BBB and absorb in the human intestine (Table 4). Results also revealed that Human Colon Adenocarcinoma cell line (Caco-2) cells may pass through all tested substances, including ETV and RPV. Drugs are metabolized by Cytochrome P450 (CYP450) isozyme, which aids in drug excretion from the body and lessens the impact of the drug. These isozymes also have a significant impact on medication interactions. NVP and RPV were the only studied substances that were anticipated to be non-substrates for CYP450 3A4. The above results are in line with the literature reports that drug-drug interaction shown by NVP and RPV are due to the induction of CYP450 3A4 enzyme. Inhibition of CYP450 isozyme results in drug-drug interaction. Most of the designed compounds were found to be non-inhibitors of these enzymes. All compounds were predicted to inhibit enzyme CYP450 1A2, except compound 4. All the tested compounds along with NVP and RPV were predicted to be non-inhibitors for enzymes CYP450 2C9, CYP450 2D6 and CYP450 3A4. The in silico results also showed these compounds are non-carcinogenic and non-mutagenic[28].
The parameters for evaluating the drug absorption are solubility and Caco-2 permeability as listed in Table 4. The solubility ranges from 2.99 to 3.99 indicating the moderate solubility of DAPYs[24,25]. The medium water solubility can be due to 1-4 polar groups present in each compound capable of forming bonds with amino acids. The Caco-2 cells permeability of 10 compounds were high. The gastrointestinal absorption of the DAPYs has shown high absorption rate. The distribution of drug candidates considers the BBB permeability. The metabolism and total clearance of the DAPYs were analysed. The DAPYs showed 2 compounds which showed Ames toxicity and hence may be mutagenic. In a collective sense, the ADMET profile of the DAPYs is satisfactory and hence suitable for in silico studies with HIV proteins.
Interactions between the docked ligands and proteins predict possible modes of action or lack thereof. The presence of hydrogen bonding in the complex formed by DAPYs and the studied RT enzyme increase the affinity of the complex and gives DAPYs a pharmacological importance. Actually hydrogen bonds play a major role in the pharmacological effect of ligands. The compounds with lower binding energy can have a higher affinity towards target proteins. These conformations are the configuration with the lowest binding autokinetic energy among the 100 conformations after compound molecules were docked with HIV-1 RT and their binding energies are -11.96 and -11.37 kcal/mol, so they are also a relatively stable binding mode. The following sections cover significant docking results and analysis.
Fig. 3 depicts the pattern of coupling between the HIV-1 RT residue and the most active molecule, the compound 3 of DAPYs derivative. The docking simulations of compound 3 with HIV-1 WT RT indicated that the binding mode resembles the approved drug RPV (fig. 4A).

Fig. 3: Binding mode of 2ZD1 with (A) Compound 3 in 3 Dimensional (3D) and (B) Compound 3 in 2 Dimensional (2D) with amino acid interactions,
( ) Conventional hydrogen bond; (
) Conventional hydrogen bond; ( ) Carbon hydrogen bond; (
) Carbon hydrogen bond; ( ) π anion; (
) π anion; ( ) π-donor hydrogen bond; (
) π-donor hydrogen bond; (  ) π-σ; (
) π-σ; ( ) π-π stacked; (
) π-π stacked; ( ) π- π T-shaped; (
) π- π T-shaped; ( ) Alkyl and (
) Alkyl and ( ) π-alkyl
) π-alkyl
In particular, compound 3 showed similar binding orientations, horseshoe conformational shape and a new hydrogen-bonding interaction between the hydrazine and drug-resistant mutants Glu138 was observed, this is why compound 3 has excellent resistance. However, the compound did not increase the interaction with the Trp229 in the hydrophobic region (fig. 3). Particularly, their common binding pattern can be described as follows. The ring Ⅰ of the compounds fits into the aromatic-rich binding sub-pocket, which is formed by the aromatic side chains of Tyr188, Tyr181 and Phe227. The 4-cyanovinyl-substituted phenyl group is parallel to the side-chain phenyl group of Tyr181, exhibiting pipi (π-π) stacking effect. Pyrimidine core in compound 3 forms two hydrogen bonds with backbone Amino (-NH), besides -NH forms another one with backbone carbonyl of residue Lys101, the oxygen atom of the Carbonyl (-CO) group of Lys101 forms one HB with the hydrogen atom of -NH groups of compound 3, which forms a -CO…NH hydrogen bond with a distance of 1.91Å. The cyanovinyl group in the ring Ⅰ of compound 3 was positioned in a cylindrical tunnel formed by highly conserved Phe227 and Trp229 (named tolerant region III). The extra 4-cyano group-substituted phenyl group incorporated to enhance the binding affinity and to optimize the inhibitors water solubility, locates around amino acids Tyr318, Leu234 and Val106, which can be well adopted in the NNIBP. Additionally, the polar hydrophilic substituent of Cyano (-CN) group resides between Val106 and Leu234 and is oriented directly toward a solvent exposure region (fig. 4A).

Fig. 4: Predicted binding models of compound 3 and TMC278 in the NNIBPs of (A) WT HIV-1 RT (PDB code: 2ZD1) and (B) Lys103/Tyr181 double mutated HIV-1 RT. Compound 3 was shown in blue and TMC278 in green. Hydrogen bonds are indicated with dashed lines in green and all the hydrogen atoms are omitted for clarity
Therefore, compound 3 probably reserves a tighter interaction with RT than other compound, which may contribute to the higher anti-HIV-1 activity of compound 3 as compared to other compounds, except compound 2.
Lys103 and Tyr181 are the two resistance mutations most frequently observed in patients treated with NNRTIs and viruses carrying these mutations show high levels of resistance to existing NNRTIs.
The effect of Tyr181 mutation on this skeleton of compound 3 shall be discussed as follows. The losing of π-π stacking effect in the interactions might be crucial for drug resistance. When docking into the Lys103/ Tyr181 mutant RT, compound 3 keep the original U-shaped conformation (fig. 4B) to maintain the activity. Despite the well-super imposition between compound 3 and RPV in molecular docking, the benzene ring Ⅰ of compound 3 was shifted a little away from the benzene ring Ⅰ of RPV for the cyanovinyl group of compound 3 to get deepens into the hydrophobic channel composed of Trp229, Tyr188 and Leu234 and get much closer to Tyr188. From theoretical speculation, compound 3 should have excellent anti-drug resistance, but the experimental result show that compound 3 is only a moderate drug-resistant inhibitor. The result could be explained by the distorted structure lose a channel and lose a hydrogen bond with Glu138 (fig. 5), the affinity loss with the sub-pocket in NNIBP to some extent, which might influence the biological activity significantly.

Fig. 5: Binding mode of 3BGR with (A) Compound 3 in 3D and (B) Compound 3 in 2D with amino acid interactions, ( ) van der Waals; ( )
Conventional hydrogen bond; ( ) π-donor hydrogen bond; ( ) π-σ; ( ) π-π stacked; ( ) π- π T-shaped; ( ) Alkyl and ( ) π-alkyl
To understand the role of molecular, steric and hydrophobic properties in the biological activity of novel class of NNRTIs, SAR studies are performed on some new DAPY derivatives. High-resolution structures of RT provide opportunities for understanding inhibitor-protein interactions with greater accuracy, more reliable determination of the structural effects of resistance mutations and systematic structure-based drug design targeting the pocket that binds to NNRTIs. The entrance to the nucleic acid-binding tunnel site suggests the possibility of extending NNRTIs so that they could interact directly with the conserved residues involved in deoxynucleotide Triphosphates (dNTPs) and/or nucleic acid binding[29]. Therefore, 2ZD1 with the best resolution is selected here as the SARs analysis object (Table 2).
The structural variety of the linker, linking the left wing and the central pyrimidine ring was the target of additional modulations. The significant SAR and analysis are covered in the following sections.
Modifications of the linker between the wing I and the central pyrimidine ring were explained here. The structural modifications on the CH2-DAPY linker, which includes a hydroxyimino group, a -CN, a Chloro (-Cl), a hydrazine and a hydroxyl substituent, were recently reported by Fen-Er Chen’s group. These new DAPY analogues were tested to see if adding hydroxyimino, -CN or other groups could enhance their antiviral activity and provide fresh building blocks for the creation of HIV-1 NNRTIs in the future[30]. The majority of the hydroxyimino analogues showed strong antiviral activity against WT virus at concentrations as low as nanomolar (Table 2). The compound with the lowest half maximal Effective Concentration (EC50) (EC50=0.005 μM) and moderate efficacy (EC50=13.23 μM) against the Lys103/Tyr181 double mutant strain was the most promising of this novel series.
Few of the -CN substituted compounds were efficacious against the double mutant strain (Lys103/Tyr181), although they were highly effective against the WT HIV-1. Compound 10 in this group demonstrated the greatest efficacy (EC50=0.059 μM). In order to help the SAR investigations to find more effective leads against HIV-1, molecular modeling studies were used to better understand the interactions between these inhibitors and HIV-1 RT.
The result also showed that the flexibility of these substituted groups can play a very important role in enhancing the activity together with the steric effects. These alkyl groups are less effective than the hydrazino group, which is probably related to the fact that the unsubstituted NH2 group could easily form a H-bond with the Val179 residue, while the substituted NH structures are less beneficial for the H-bond formation, although the alkyl group could occupy more space in the Val179 pocket. This is why compound 3 has better activity and lowest free energy of binding than the other 9 compounds.
Introducing a hydrazine group, might improve the hydrogen-bonding interactions of these inhibitors with RT. Apparently, as designed, the hydrazine group on the methylene linker of hydrazine-CH2-DAPY enhanced the interaction with HIV-1 RT in WT. The observed high effectiveness against HIV-1 WT, however, is explained by molecular modeling, which indicates that compound 3 lacks interaction with the important mutant amino acid residues of Asn103 in the NNIBPs, but weak activity against the double-mutant strain RES056 (Lys103/Tyr181)[6]. On the other hand, either a hydrazine or hydroxyl groups has also been proven to be very beneficial for improving the antiviral activity, especially increasing the drug resistance activities, in particular, compound 5 had the best resistance. It has already been shown that there are still some possibilities to achieve the anti-drug resistance effects by modifying the linker group between the left benzene ring and the central pyrimidine ring.
Modifications of the wing I is explained here. Of these compounds 6, 7 and 9 have the same substituents at different positions of phenyl ring I, whereas 1, 4, 5 and 10 are substituted by different groups at the same position.
The relationship between the conserved amino acid Trp229 in NNIBP and the addition of a -CN group to DAPYs was recognized to increase both biological activity and drug resistance. By restricting the flexible rotation of the phenyl ring I of DAPYs, a 2-substituted group on the phenyl ring of DAPYs with higher-stacking between the ligand and Y181 or Y188 in the hydrophobic pocket of NNIBP can help this type of contact[1]. It was also observed that most of the active DAPY derivatives shared both 4-CN group and 2-substituted groups. For example, TMC125 contains two methyl groups on the 2, 6-positions of the left phenyl ring (fig. 1)[31]. The possibility for the insertion of 4-substituted group into the Trp229-containing narrow pocket of NNIBP is dependent on both the physical properties (including the steric effects etc.) of the 4-substituted group and the synergistic effect of the C2-substituted groups on the phenyl ring. In some circumstance, the function of the C2-substituted groups might exert much more assisting effect than the 4-substituted group via fixing the suitable conformations of the 4-substituted group. The 4-cyanovinyl could adjust its conformation to adapt with the hydrophobic channel by Sigma (σ)-bond rotation, while the -CN group could exert a similar effect only by the assistance of the 2-substituted groups.
The introduction of the cyanovinyl group could enhance the biological potency against WT virus strain, while the same group could also decrease the cytotoxicity, which again verified the cyanovinyl group at 4th position to be superior to other substituents (compound 3). Based on this analysis, we envisioned that by substitution with aromatic rings, replacements of 4-cyanovinyl could satisfy the spatial orientation of the well tolerated cylindrical hydrophobic tunnel (tolerant region III) and yield more optimal aryl-aryl hydrophobic interactions. Compound 3’s addition of a cyanovinyl group raised its antiviral activity (EC50), most likely as a result of improved interactions between the cyanovinyl group and Try18.1, Try188, Phe227 and Trp229 residues. The results have shown that both the -CN and the cyanovinyl groups are beneficial for enhancement of the anti-HIV activity.
In terms of SAR, sterically small substituents, such as –CH2 (8), -Cl (6, 7) and (Bromo (-Br)) (1, 2) in the 2 or 4 positions, which were well tolerated were bind to the side chains of Tyr181 and Tyr188 of HIV- 1 RT. The SAR found that halogenated hybrids (-Br) had improved anti-HIV-1 activity, which may be because of the addition of halogen atoms which made the hybrids more lipophilic. According to the SAR, hybrids with 2-Cl, 2-F and 2-Br substituents exhibited considerably increased activity against the WT HIV-1 strain (compound 10), whereas the activity against the WT HIV-1 strain was lowered by the electron-donating groups at 2-Methoxy (2-OCH3).
Also, according to the SAR, hybrids with 2-Cl, 2-F and 2-Br substituents exhibited considerably increased activity against the WT HIV-1 strain, whereas the activity against the WT HIV-1 strain was lowered by the electron-donating groups at 2-OCH3. A preliminary SAR analysis of these target molecules highlighted a preference for the smaller or more flexible group’s substitution in the linker between the left ring and the pyrimidine skeleton for enhancing the anti-HIV activity. As shown in fig. 6, the 2nd position is the most preferred position to improve antiviral activity and the introduction of the cyanovinyl group probably enhanced interactions of the cyanovinyl group with Tyr181, Tyr188, Phe227 and Trp229 residues, which is favorable for improvement of water solubility and other drug-like properties. The cyanovinyl C4-substituent significantly improved the potency against WT HIV-1 and several mutant strains.

Fig. 6: A systematic summary of SAR
In this study, the interaction between HIV RT and DAPY molecules was stimulated by molecular docking, show that the binding mechanism between DAPY derivatives and RT protease was expounded. This work might provide useful information for guiding the rational design of potential HIV-1 NNRTI DAPYs. DAPYs superior pharmacological characteristics and distinctive molecular binding method will keep medicinal chemists motivated to discover additional effective HIV-1 NNRTIs.
Acknowledgements:
This research was supported by the Natural Science Foundation of Xinjiang Uygur Autonomous Region (2022D01A18).
Conflict of interests:
The authors declared no conflict of interest.
References
- Thakur A, Thakur M, Bharadwaj A, Thakur S. SAR and QSAR studies: Modelling of new DAPY derivatives. Eur J Med Chem 2008;43(3):471-7.
[Crossref] [Google scholar] [PubMed]
- Sun L, Gao P, Dong G, Zhang X, Cheng X, Ding X, et al. 5-Hydroxypyrido [2, 3-b] pyrazin-6 (5H)-one derivatives as novel dual inhibitors of HIV-1 reverse transcriptase-associated ribonuclease H and integrase. Eur J Med Chem 2018;155:714-24.
[Crossref] [Google scholar] [PubMed]
- Zhou Z, Liu T, Kang D, Huo Z, Wu G, Daelemans D, et al. Discovery of novel diarylpyrimidines as potent HIV-1 NNRTIs by investigating the chemical space of a less explored “hydrophobic channel”. Org Biomol Chem 2018;16(6):1014-28.
- Namasivayam V, Vanangamudi M, Kramer VG, Kurup S, Zhan P, Liu X, et al. The journey of HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) from lab to clinic. J Med Chem 2018;62(10):4851-83.
[Crossref] [Google scholar] [PubMed]
- Prajapati DG, Ramajayam R, Yadav MR, Giridhar R. The search for potent, small molecule NNRTIs: A review. Bioorg Med Chem 2009;17(16):5744-62.
[Crossref] [Google scholar] [PubMed]
- Lehman DA, Wanalwa DC, McCoy CO, Matsen FA, Langat A, Chohan BH, et al. Low-frequency nevirapine resistance at multiple sites may predict treatment failure in infants on nevirapine-based treatment. J Acquir Immune Defic Syndr 2012;60(3):225-33.
[Crossref] [Google scholar] [PubMed]
- Huang B, Li C, Chen W, Liu T, Yu M, Fu L, et al. Fused heterocycles bearing bridgehead nitrogen as potent HIV-1 NNRTIs. Part 3: Optimization of [1, 2, 4] triazolo [1, 5-a] pyrimidine core via structure-based and physicochemical property-driven approaches. Eur J Med Chem 2015;92:754-65.
[Crossref] [Google scholar] [PubMed]
- Lansdon EB, Brendza KM, Hung M, Wang R, Mukund S, Jin D, et al. Crystal structures of HIV-1 reverse transcriptase with etravirine (TMC125) and rilpivirine (TMC278): Implications for drug design. J Med Chem 2010;53(10):4295-9.
[Crossref] [Google scholar] [PubMed]
- Kang D, Feng D, Ginex T, Zou J, Wei F, Zhao T, et al. Exploring the hydrophobic channel of NNIBP leads to the discovery of novel piperidine-substituted thiophene [3, 2-d] pyrimidine derivatives as potent HIV-1 NNRTIs. Acta Pharm Sin B 2020;10(5):878-94.
[Crossref] [Google scholar] [PubMed]
- Zhan P, Chen X, Li D, Fang Z, de Clercq E, Liu X. HIV‐1 NNRTIs: Structural diversity, pharmacophore similarity and implications for drug design. Med Res Rev 2013;33(S1):E1-72.
[Crossref] [Google scholar] [PubMed]
- de Béthune MP. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development and use in the treatment of HIV-1 infection: A review of the last 20 y (1989-2009). Antiviral Res 2010;85(1):75-90.
[Crossref] [Google scholar] [PubMed]
- Feng XQ, Zeng ZS, Liang YH, Chen FE, Pannecouque C, Balzarini J, et al. Synthesis and biological evaluation of 4-(hydroxyimino) arylmethyl diarylpyrimidine analogues as potential non-nucleoside reverse transcriptase inhibitors against HIV. Bioorg Med Chem 2010;18(7):2370-4.
[Crossref] [Google scholar] [PubMed]
- Ma XD, Yang SQ, Gu SX, He QQ, Chen FE, de Clercq E, et al. Synthesis and anti‐HIV activity of Aryl‐2‐[(4‐cyanophenyl) amino]‐4‐pyrimidinone hydrazones as potent non‐nucleoside reverse transcriptase inhibitors. ChemMedChem 2011;6(12):2225-32.
[Crossref] [Google scholar] [PubMed]
- Meng G, Liu Y, Zheng A, Chen F, Chen W, de Clercq E, et al. Design and synthesis of a new series of modified CH-diarylpyrimidines as drug-resistant HIV non-nucleoside reverse transcriptase inhibitors. Eur J Med Chem 2014;82:600-11.
[Crossref] [Google scholar] [PubMed]
- Liu Y, Meng G, Zheng A, Chen F, Chen W, de Clercq E, et al. Design and synthesis of a new series of cyclopropylamino-linking diarylpyrimidines as HIV non-nucleoside reverse transcriptase inhibitors. Eur J Pharm Sci 2014;62:334-41.
[Crossref] [Google scholar] [PubMed]
- Gu SX, He QQ, Yang SQ, Ma XD, Chen FE, de Clercq E, et al. Synthesis and structure-activity relationship of novel diarylpyrimidines with hydromethyl linker (CH (OH)-DAPYs) as HIV-1 NNRTIs. Bioorg Med Chem 2011;19(17):5117-24.
[Crossref] [Google scholar] [PubMed]
- Gu SX, Li ZM, Ma XD, Yang SQ, He QQ, Chen FE, et al. Chiral resolution, absolute configuration assignment and biological activity of racemic diarylpyrimidine CH (OH)-DAPY as potent non-nucleoside HIV-1 reverse transcriptase inhibitors. Eur J Med Chem 2012;53:229-34.
[Crossref] [Google scholar] [PubMed]
- Yan ZH, Huang XY, Wu HQ, Chen WX, He QQ, Chen FE, et al. Structural modifications of CH (OH)-DAPYs as new HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg Med Chem 2014;22(8):2535-41.
[Crossref] [Google scholar] [PubMed]
- Yan ZH, Wu HQ, Chen WX, Wu Y, Piao HR, He QQ, et al. Synthesis and biological evaluation of CHX-DAPYs as HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg Med Chem 2014;22(12):3220-6.
[Crossref] [Google scholar] [PubMed]
- Li TT, Pannecouque C, de Clercq E, Zhuang CL, Chen FE. Scaffold hopping in discovery of HIV-1 non-nucleoside reverse transcriptase inhibitors: From CH (CN)-DABOs to CH (CN)-DAPYs. Molecules 2020;25(7):1581.
[Crossref] [Google scholar] [PubMed]
- Daina A, Michielin O, Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 2017;7(1):1-3.
[Crossref] [Google scholar] [PubMed]
- Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, et al. admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model 2012;52:3099-105.
[Crossref] [Google scholar] [PubMed]
- Morris GM, Huey R, Olson AJ. Using AutoDock for ligand‐receptor docking. Curr Protoc Bioinformatics 2008;24(1):8-14.
[Crossref] [Google scholar] [PubMed]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 1997;23(1):3-25.
[Crossref] [Google scholar] [PubMed]
- Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 2002;45(12):2615-23.
[Crossref] [Google scholar] [PubMed]
- van de Waterbeemd H, Gifford E. ADMET in silico modelling: Towards prediction paradise? Nat Rev Drug Discov 2003;2(3):192-204.
[Crossref] [Google scholar] [PubMed]
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 2009;30(16):2785-91.
[Crossref] [Google scholar] [PubMed]
- Lubomirov R, Csajka C, Telenti A. ADME pathway approach for pharmacogenetic studies of anti-HIV therapy. Pharmacogenomics 2007;8:623-33.
[Crossref] [Google scholar] [PubMed]
- Nanni RG, Ding J, Jacobo-Molina A, Hughes SH, Arnold E. Review of HIV-1 reverse transcriptase three-dimensional structure: Implications for drug design. Perspect Drug Discov Des 1993;1(1):129-50.
- Zhuang C, Pannecouque C, de Clercq E, Chen F. Development of non-nucleoside reverse transcriptase inhibitors (NNRTIs): Our past twenty years. Acta Pharm Sin B 2020;10(6):961-78.
[Crossref] [Google scholar] [PubMed]
- Das K, Clark AD, Lewi PJ, Heeres J, de Jonge MR, Koymans LM, et al. Roles of conformational and positional adaptability in structure-based design of TMC125-R165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J Med Chem 2004;47(10):2550-60.
[Crossref] [Google scholar] [PubMed]





